

You might say this article culminates the two years this site has been on the web.
It is about treatment of the most common stone patients, treatment to prevent more stones, and therefore the topmost important matter for patients and their physicians.
The topic is so important I plan three versions. This one is primary, and has not only references but linked documents so original materials are available to everyone. The next will be a video that offers the material in a more fluent if less documented format. Finally Jill Harris has promised to coauthor with me a version in her lovely and popular style.
I present all of the treatment trials in the context of the supersaturation hypothesis for stone formation and in the light of what we know about urine stone risk factors. The entire site thus far concerns these matters, and was in fact created to support this article which is the capstone of the site to date.
This article draws importantly on three other articles that describe the kidney stone treatment diet. As things have turned out, the 2015 – 2020 US recommended diet more or less exactly matches the needs for stone prevention in idiopathic calcium stone formers. The scientific reasons for this are well established. Therefore this article is meant to be read in parallel with the above three diet articles.
It also draws on our articles on thiazide, potassium citrate, idiopathic hypercalciuria, the idiopathic calcium stone forming phenotypes, and regulation of oxalate excretion, and these links are embedded in the text.
The beautiful picture is Hidden Lake, Bearhat Mountain, Glacier N.P., near Kalispell, Montana. I had thought to use Ansel Adams but this one is by Howie Garber, a modern professional and I think wonderful photographer who had this print on his site.
WHO ARE IDIOPATHIC CALCIUM STONE FORMERS?
They are people who form calcium stones in the absence of any causal systemic disease. I have listed the more common such diseases in another article. Of course, this requires stone analyses be done, hopefully for all or a large majority of stones from a patient. Patients whose stones contain uric acid, cystine, struvite or drug crystals are not included here, being special cases.
Although the idiopathic calcium stone formers come in three separable groups, for the purposes of prevention all three are treated much the same way, with a few exceptions. The most common, calcium oxalate stone formers, have certainly predominated in all of the trials by force of numbers. The hydroxyapatite calcium phosphate stone formers have surely been part of most trial groups but have not been reported separately. The uncommon brushite stone formers may or may not have been in trials but are scarce enough their numbers would be too slight to alter final results and their particular outcomes from a given treatment impossible to gauge looking back. Therefore I do not exclude brushite stone formers having no real reason to do so.
THEORETICAL STONE RISK FACTORS
Determinants of Urine Supersaturation
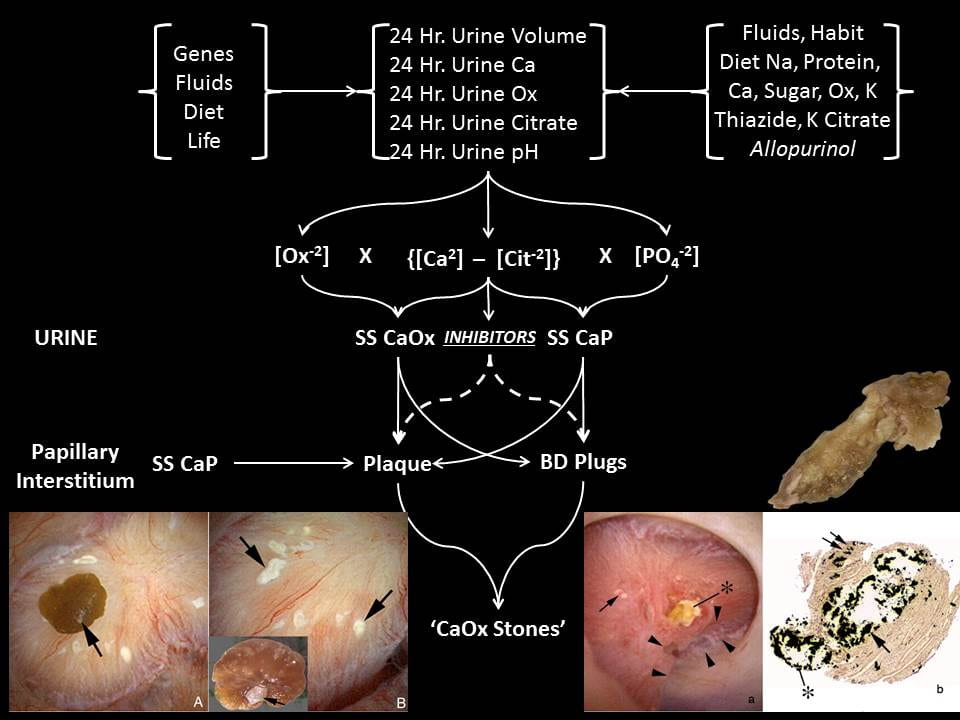
In a schematic sense, the two relevant supersaturations SS CaOx and SS CaP are approximated by the products of calcium and oxalate and calcium and divalent phosphate ion, respectively. The calcium free to combine with oxalate or phosphate is crudely the difference between the calcium and citrate concentrations in mEq/l, because citrate binds calcium in a soluble complex. The divalent phosphate is controlled by the
phosphate molarity and urine pH – higher pH, higher abundance of the divalent form. The oxalate is mostly just the oxalate concentration.
 EQUIL 2, the common computer program used to calculate supersaturations, in fact calculates in this way but considers all possible ion associations.
EQUIL 2, the common computer program used to calculate supersaturations, in fact calculates in this way but considers all possible ion associations.
Therefore the main factors causing stones via supersaturation are the excretions of water, calcium, oxalate, and citrate, and urine pH (Figure at left). These are controlled by genes (idiopathic hypercalciuria and perhaps oxalate and citrate and pH), fluid intake, diet oxalate, calcium, sodium, and acid load. Over it all is life itself that determines not only the daily totals but their variability. We measure using 24 hour urine collections, but relative excretions of water, calcium, oxalate, and citrate and urine pH change throughout the day, so supersaturation may have many peaks and valleys.
Inhibitors – The Inaccessible Quotient
Molecules that Inhibit Crystals
It is not so much that we do not know any urine crystallization inhibitors as it is we can name too many. Urine contains about 1,800 identifiable proteins among which are many anionic species easily able to attach to calcium based crystals and slow their growth or even nucleation. The link concerns stone associated proteins as they are more likely than not to have attached to crystals. The proteins that matter in terms of stone forming propensity, hundreds, perhaps, lie hidden amongst the hordes of proteins, so we do not know which proteins really have large effects and are important for us to measure.
Citrate stands out as the one we can and do measure. It can disrupt calcium crystal growth at concentrations far below those in urine. Just as the unbound calcium is that free to create crystals, the unbound citrate is free to inhibit crystals, so the calcium – citrate molar difference would be predicted as a critical factor in inhibition.
Citrate and The Ostwald Limit
Wilhelm Ostwald was born on September 2, 1853, in Riga, Latvia. Among his famous pupils are Arrhenius (Nobel Prize 1903),Van ‘t Hoff (Nobel Prize 1901), Nernst (Nobel Prize 1920) – taken from the Nobel Lectures. He won his Nobel in 1909 and died in 1932.
The Ostwald limit is the supersaturation at which crystallization begins in a given solution. It can be measured in human urine, and if supersaturation is the floor the limit is the ceiling. That distance, between supersaturation and the limit is smaller in stone forming patients than in normal people, especially for calcium phosphate. Variation of urine citrate concentration in the range found in urine of between 0.5 and 5 mmol/l seems to account for 1/2 of the elevation of the limit above supersaturation. Therefore as a risk factor and as a treatment citrate ought to be very important and perhaps mysterious, too, for it affects supersaturation – via calcium binding, inhibits those crystals supersaturation would produce, and therefore has a role in setting the upper limit of metastable supersaturation – that privileged space between supersaturation and the limit in which a solution remains clear of crystals.
Anchored Tissue Calcifications
Most stones form over anchored tissue calcifications: Interstitial HA crystals, plaque, and terminal collecting duct of Bellini (BD) plugs.
Plaque
On the left of the intra-operative image panel that is itself at the bottom left of the figure above you can see white plaque with a CaOx stone growing on it (arrow) – the removed stone (inset) has a central white CaP anchor site which attached this stone to the plaque. The big arrowhead points to where it was attached. I have made a video that may help clarify this process. Urine CaP SS can create the initial layer over interstitial plaque that becomes exposed to urine when the urothelial boundary gives way.
Plaque itself presumably arises from supersaturations in the papillary interstitium, not the urine, and these cannot be measured at the present time. Some theory exists and has been partially tested. It suggests that idiopathic hypercalciuria may foster plaque but this idea is too untested to affect clinical practice.
Plugs
Urine CaP SS can cause plugging of terminal collecting ducts. A long plug is shown in the right lower panel of the above figure. The shaft is HA and filled the duct. The rounded end arose from the urine over the exposed plug. The OVergrowth can be CaOx stones, CaP stones, or both depending on the urine supersaturation balance that is conditioned by the five factors I have already mentioned: volume, calcium, oxalate, and citrate excretions, and urine pH.
THE SUPERSATURATION HYPOTHESIS
The Separate Roles of CaP and CaOx SS and of Citrate
Urine CaP SS is what must drive formation of the initial HA overgrowth on plaque, and urine CaOx SS must drive subsequent formation of the CaOx that makes up the bulk of the stone. Urine CaP SS is what must drive plugging of BD, and urine CaOx and CaP SS will then determine the relative amounts of these two crystals over the open end of the plug facing into the urine. Urine citrate is crucial in two ways. By binding calcium it lowers both CaP and CaOx SS. By inhibiting CaP and CaOx SS it contributes to the ULM for each and presumably protects against overgrowth on plaque, plugging, and overgrowth on plugs. Since only free citrate can so act, the molar difference between calcium and citrate ions is presumably a crucial determinant of the inhibition effect. This latter point has not been critically tested. Apart from urine citrate, we do not know enough about inhibitors and local tissue factors to use them in stone prevention, but we do have a powerful prediction from the physical chemistry of crystallization.
The Clinical Meaning of the Supersaturation Hypothesis
GIven that stones depend on crystal production, and given that supersaturation is a necessary factor in crystal production, one can say that: The urine supersaturations of an active stone former are too high in relation to the crystals in stones forming. By too high I mean simply high enough that, all things considered – inhibitors, tissue regulation of crystallization – crystals are being formed. Because CaP SS may indeed have a dual role – in forming the plaque attachment site for CaOx stones and being the main crystal in plugs, even when stones have little HA one might conclude the CaP SS is too high.
One immediate deduction from this formulation is that prevention of new stones requires that urine supersaturations with respect to the crystals in stones forming be lowered, and that lowering needs to come from alterations of those urine factors that control supersaturation. As well, even when stones are CaOx, CaP SS should be lowered.
Another deduction is that empirical urine stone risk factors will in fact be mainly those very factors that most influence SS: urine calcium, oxalate, citrate, volume, and pH.
EMPIRICAL STONE RISK FACTORS
Elsewhere I have presented the important work of Gary Curhan, and do so again here. They are in fact those predicted from the primacy of supersaturation because all of them affect supersaturation in a direct manner.
Urine Calcium
 The light red and blue (female and male) bars are plotted downward from the relative risk of becoming a stone former to the lower 95th percentile of that relative risk. The solid bars fill in the space between the end of that 95th percentile and 1, the point of no relative risk increase.
The light red and blue (female and male) bars are plotted downward from the relative risk of becoming a stone former to the lower 95th percentile of that relative risk. The solid bars fill in the space between the end of that 95th percentile and 1, the point of no relative risk increase.
For urine calcium (upper left plot) relative risk with respect to less than 100 mg/day rises progressively in the two female and one male cohort. Between 100 – 149 mg/day the lower 95th percentile crosses 1 meaning the relative risk it not significantly above 1. In the next hexile relative risk is almost certainly significant and thereafter it certainly is. Of crucial importance there is no knife edge; urine calcium is a smoothly graded risk factor.
Urine Oxalate
Urine oxalate behaves similarly except the magnitude of relative risk (with respect to <20 mg/d) does not rise as far as it does for calcium excretion.
Urine Citrate
Urine citrate opposes supersaturation, so one expects that relative risk will fall as its excretion rises, which is a well known clinical maxim that the Curhan data gives a proper experimental foundation. In this case relative risk is plotted upward from below 1 in most cases, and the upper 95th percentile is in the red and blue solid bars. Significant risk exists at values below 400 mg/day. Notably the change in risk seems modest compared to that of urine calcium or oxalate, but partly that is because the available range is only from 1 to 0. Perhaps a lot of the protection from citrate arises from direct crystal inhibition which is rather complete at concentrations below those in urine. I have narrowed the y axis range to make the progression more obvious.
Urine Volume
Urine volume is like citrate; relative risk falls markedly as volume rises, but in some cohorts the upper bound of risk ratios are above 1 until volume reaches above 2 liters – solid bars above 1. I have narrowed the vertical axis range to make the progression of risk reduction more obvious.
Urine pH and Other Factors
Urine pH would have a predictably complex relationship to stone risk, in that low values would cause uric acid stones and high values CaP stones. So one might have predicted a U shaped curve. In fact, if you inspect table 3 from his main paper such a pattern is perhaps present in the first of the female cohorts, relative risk (with respect to <5.5) fell between 5.5 and 6 then rose again; this was also true in the second female cohort but not the male cohort in which risk rose with urine pH.
Urine phosphate is very plentiful compared to oxalate or calcium so pH would presumably control the abundance of the divalent form and therefore stone risk. In two of the three cohorts relative risk (as compared with <600 mg/d) was higher at higher phosphate excretions.
I showed observational data decades ago that reducing urine uric acid excretion with allopurinol reduced calcium oxalate stones, and a double blind randomized trial substantiated this fact, but in the Curhan studies stone risk did not rise and, in several cohorts fell as urine uric acid increased.
These data all arise from one publication. Here is a link to a spreadsheet with all of the data I extracted from Table 3 of the paper. Links to the paper online and to a PDF are in the spreadsheet.
THE SPECIAL CASE OF URINE URIC ACID
Long ago, I published observations from my clinic that some calcium oxalate stone formers excreted a lot of uric acid in their urine (>750 mg/d and >800 mg/d women and men, respectively) and I had decided on purely clinical instincts to see of lowering it with allopurinol might reduce new stone production. Among 21 patients observed for an average of 1.85 years only one had a single recurrent stone, whereas prior to treatment all had been very active stone formers.
The work was meant only to begin a conversation about the possibility that uric acid had some role in calcium oxalate stone formation, and the paper called for a proper trial to determine if the matter was worth pursuing.
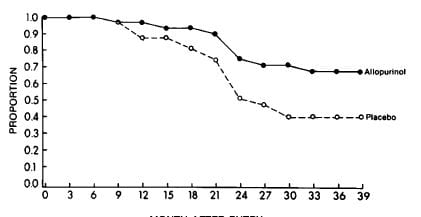 Bruce Ettinger performed such a trial. In a randomized, prospective double blind trial lasting 39 months 31 placebo treated patients had 18 new stones and 29 allopurinol treated patients had 9 stones (X2=3.4 p=0.035). His life table analysis showed a highly significant (p<0.01) treatment effect with 42% placebo stone free and 69% treated stone free.
Bruce Ettinger performed such a trial. In a randomized, prospective double blind trial lasting 39 months 31 placebo treated patients had 18 new stones and 29 allopurinol treated patients had 9 stones (X2=3.4 p=0.035). His life table analysis showed a highly significant (p<0.01) treatment effect with 42% placebo stone free and 69% treated stone free.
Decades of bench research have failed to provide a mechanism by which these treatment effects might be brought about. So we have reasonable empirical evidence for an phenomenon whose cause is not known. I mention allopurinol as a treatment alternative in later sections of this article but do not pool the allopurinol trial with those for diet, thiazide, or potassium citrate.
RISK FACTOR MODERATION REDUCES STONES
Our evidence arises from such stone prevention trials we have available, and although they do not satisfy professional trialists they satisfy me as a scientist. Their message – treatments work – is very unlikely to be wrong.
I have compiled the main data for all of the trials. Here is a link to the spreadsheet of the data. On the sheet are links to PDF images of the publications as well as PubMed links.
Water for Stone Prevention
In patients who had formed a first stone, Borghi (1996) compared a high water intake to minimal treatment. Stones appear to have been mainly 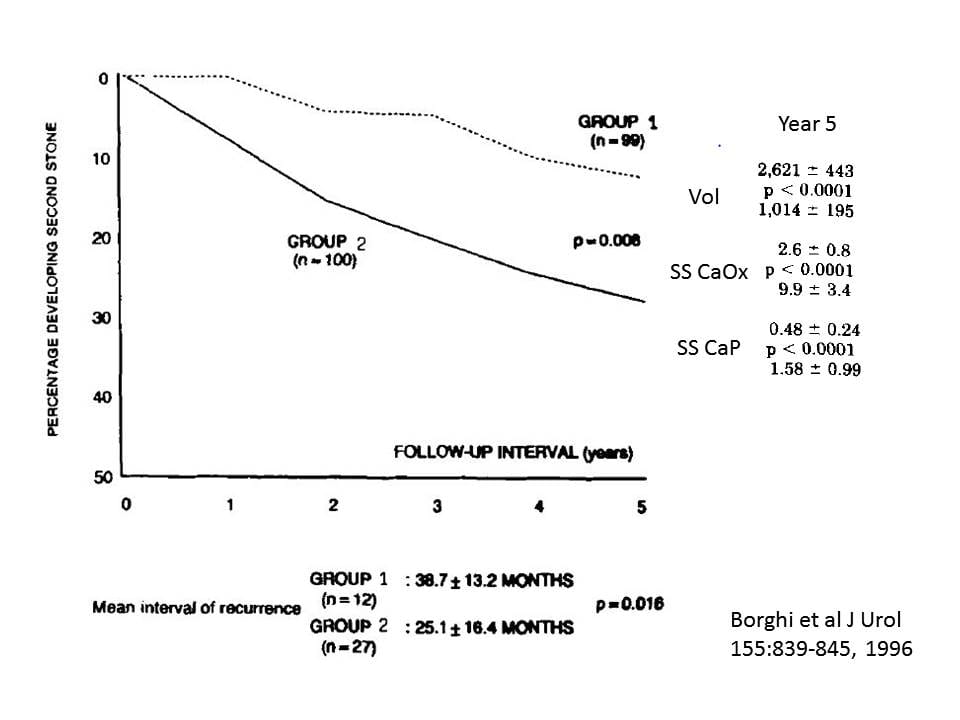 CaOx. Patients with hypertension, retained stone fragments in their kidneys, or ‘other metabolic pathology’ were excluded.
CaOx. Patients with hypertension, retained stone fragments in their kidneys, or ‘other metabolic pathology’ were excluded.
Of note, these patients had low baseline urine volumes (ml/d): 1,057 vs. 1,401 and 990 vs.1,239, patients vs. controls, male and female, respectively, P<0.001 for comparison of patients to controls within sex. This baseline urine volume would carry a high stone risk using the Curhan findings shown above. Urine calcium averaged 244 and 266 mg/day in Groups 1 and 2, respectively, also posing risk. The sex ratios in the two groups were about the same – 60-70% males; average age was 41 years.
Whereas the treated group was aggressively encouraged to raise urine volume, measure volume at home, and bring in a collection every year, the controls were told that ‘…since it was an isolated stone episode, it was not necessary at least at that time to follow any special procedures.’
At the end of five years, 12 of the 99 patients who raised their urine volume had formed a new stone vs. 27 of the 100 who did not (X2 = 6.9, p=0.008). The time to first stone was longer in the high fluid group as noted below the figure.
The urine SS values for CaOx and CaP were much lower in the treated vs. the placebo group as well (Year 5 data shown).
This trial indicates that at least for first time calcium stone formers a urine volume of about 2.6 liters daily will reduce but not abolish new stone formation. This is consistent with the supersaturation hypothesis as the treatment group had both less stones and lower SS than the controls. It also fits well with the Curhan data, in that less stones occurred when urine volume was above 2.5 liters.
Diet for Stone Prevention
Diet Sodium
If you have read about idiopathic hypercalciuria, the main reason calcium stone formers have rather generous urine calcium excretion rates, you know that diet sodium and protein are powerful controllers of urine calcium losses, and that diet calcium cannot be low for long without risk of bone disease.
I and Elaine Worcester compiled these data from a mixture of publication. This spreadsheet contains the data I  used to make this picture along with links to the PDF images of the articles.
used to make this picture along with links to the PDF images of the articles.
In the figure, red are normals, blue IH, circles are experimental and triangles observational data.
The message is clear: Urine calcium is powerfully influenced by diet sodium intake – which is urine sodium in the stable state. IH is, howsoever achieved, a condition in which urine calcium is unduly dependent on diet – and therefore urine – sodium.
That the slopes really do differ was tested in a general linear model with urine calcium as dependent and urine sodium and type – normal vs. IH – as independent. The slopes differed by type (cross product p<0.0001). The slope for IH was 1.396 vs. 0.380 for normals). The overall regression captured 75.6% of the variance around urine calcium. The effect of type on the intercept of the two regressions was NS.
That sodium dependence of calcium excretion is so much more dependent on sodium in IH than normal must reflect mechanisms for the pathogenesis of IH. In my detailed analysis of IH, I show, using data Elaine Worcester and I obtained, a similarly increased dependence of urine calcium on filtered load of calcium, all pointing to reductions of tubule calcium transport.
Diet Protein
Increasing diet protein intake will raise urine calcium excretion. At one time it was thought that protein increased urine calcium by imposing an acid load, but that has proven untrue. Given with enough alkali to offset the acid load protein increased urine calcium. The mechanism of protein induced hypercalciuria is a mixture of increased glomerular filtration and reduced tubule calcium reabsorption. I have reviewed this problem in a another detailed article.
Diet Calcium
Because idiopathic hypercalciuria is so common among idiopathic calcium stone formers and can result in bone mineral loss, a high calcium diet is generally recommended. Such a diet has another value in that high diet calcium reduced oxalate absorption and therefore urine oxalate excretion.
A Low Sodium and animal protein, High Calcium Diet Reduces Stones
Only one trial has concerned reduced diet sodium and it has the fault and benefit of being more inclusive in that diet protein and calcium were also modified. Note that ‘high’ and ‘low’ refer to diet changes from current habits. As detailed in multiple other articles on this site, US government recommendations now favor 1,200 mg calcium, less than 2,300 mg sodium, less than 10% total carbohydrates as refined sugar, 0.8 mg protein/kg body weight, and 112 mEq/day diet potassium from fruits and vegetables. By current standards this is a high calcium, low sodium, low sugar, low protein, high potassium diet.
The 120 subjects all were men with idiopathic hypercalciuria diagnosed by the older research criterion of above 300 mg of urine calcium daily in the absence of a systemic disease. All had at least two CaOx stones.
The trial compared a low calcium diet to a low sodium, controlled protein, high  calcium diet, 60 men in each diet arm. The low sodium diet was 50 mEq/day sodium, a low value, 1,200 mg (30 mmol) of calcium, and 93 gm of protein, 50 gm from animals and 40 gm from plant sources, and oxalate of 200 mg daily – not a low oxalate diet. The contrast group ate a low calcium diet – 400 mg/day (10 mmol) and were told to avoid high oxalate foods. Sodium and protein were not specified. By the end of five years, 12 men in the low sodium group and 23 men in the low calcium diet group had formed at least one new stone (X2 = 4.88, p=0.027).
calcium diet, 60 men in each diet arm. The low sodium diet was 50 mEq/day sodium, a low value, 1,200 mg (30 mmol) of calcium, and 93 gm of protein, 50 gm from animals and 40 gm from plant sources, and oxalate of 200 mg daily – not a low oxalate diet. The contrast group ate a low calcium diet – 400 mg/day (10 mmol) and were told to avoid high oxalate foods. Sodium and protein were not specified. By the end of five years, 12 men in the low sodium group and 23 men in the low calcium diet group had formed at least one new stone (X2 = 4.88, p=0.027).
Reduced Stones Associates with Reduced Stone Risk Factors
Individual Factors
Urine calcium levels were not different, presumably because the higher calcium intake was opposed by low sodium diet. But urine oxalate was lower in the high calcium low sodium group: 422, 411, 422, 433, 411 vs. 344, 322, 333, 333, 333 umol/day years 1 – 5, low calcium vs low sodium diet; all 5 differ as to change from baseline. So oxalate induced risk was lower. Urine volumes were the same – all about 2 liters/day or more. Urine citrate was not presented.
Low diet sodium is the likely reason a high calcium diet resulted in urine calcium levels no higher than found on a low calcium diet, and the high calcium diet presumably lowered urine oxalate as high calcium in food is known to do. So low sodium diet presumably lowered urine calcium and permitted a high calcium intake which will tend to lower urine oxalate.
Supersaturation
Only CaOx SS is presented and not in an ideal form. It was measured using EQUIL at baseline and one week but thereafter calculated by a regression formula. Even so, it was lower in the high calcium low sodium diet group: 7.3, 6.8, 6.7, 4.8, 4.7 vs. 5.1, 4.7, 4.5, 3.7, 3.5, years 1-5, low calcium vs. low sodium diet, respectively; all five differ as to change from baseline. So, despite some weaknesses, SS as estimated was lower.
Diet Oxalate
It seems odd that we have trial data on sodium, calcium, and protein but not on diet oxalate, Nevertheless to date no trial has been mounted. I believe it will fail unless the trial incorporates both high calcium intake and reduced diet oxalate together, because one needs to reach diet oxalate  levels of under 100 mg daily unless diet calcium is high.
levels of under 100 mg daily unless diet calcium is high.
Evidence for this statement is a set of studies I have summarized on this one graph. The symbols are scaled to the diet oxalate intake between 200 to 50 mg/d, and the studies are labelled by who I think of the main investigator involved. The original data are in the linked spreadsheet.
More or less independent of diet oxalate (size of symbols) urine oxalate (y axis) falls with calcium intake (x axis) and by about 1,000 mg calcium intake values more or less pack between 25 – 35 mg/d. In the small upper right box, Hess have a diet of 2,000 mg oxalate with 1,000 mg calcium – upper left point at 80 mg/d of urine oxalate, which fell to 30 mg/d when diet calcium was raised to 4,000 mg/day. The point of this exercise is that even the most remarkable oxalate intake can be balanced by a correspondingly high calcium intake.
Medication for Stone Prevention
Thiazide and Potassium Citrate
These treatments are aimed at reducing supersaturation and, in the case of citrate, increasing urine levels of a powerful inhibitor of crystallization. Both agents reduce new stones, although the trials are perhaps less that perfectly ideal. I have already mentioned the spreadsheet that has all of the key data. Likewise I have presented these trials in a more detailed fashion in two other articles – Thiazide and Potassium citrate.
In the figure, triangles are randomized, prospective, double blind trials, circles are not double blind, and size is scaled to time. The largest symbols 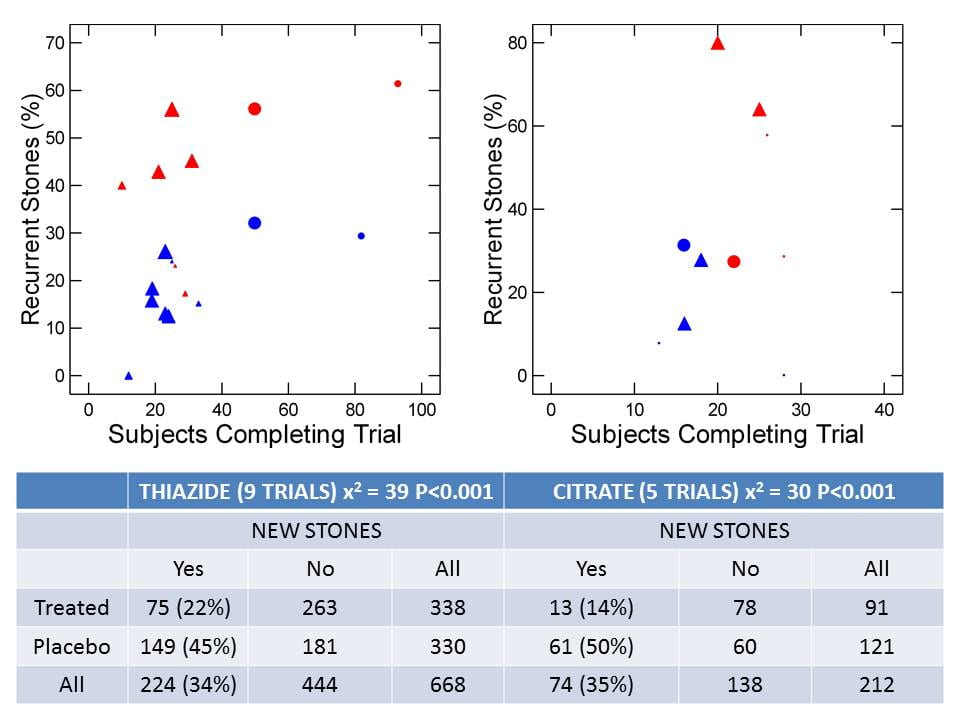 represent 3 years, the next size 2 years, the third one year and tiny points barely visible less than a year. These latter concern formation of new stones after surgery as judged radiographically. Red are control and blue treatment arms. Two randomized double blind thiazide trials had one control for two treatment arms, thus the five blue and three red large triangles.
represent 3 years, the next size 2 years, the third one year and tiny points barely visible less than a year. These latter concern formation of new stones after surgery as judged radiographically. Red are control and blue treatment arms. Two randomized double blind thiazide trials had one control for two treatment arms, thus the five blue and three red large triangles.
The vertical axis plots the fraction of patients who concluded the trial that formed a new stone, against the number of subjects who completed the trial on the horizontal axis.
The table sums up all 9 thiazide and all five citrate trials in terms of numbers of subjects finishing and number with and without new stones.
A total of 668 patients finished the 9 thiazide trials. Of these 22% formed a new stone. Among placebo patients 45% relapsed; the p value for a simple X2 test is shown. Corresponding values for the 5 citrate trials give a similar picture.
Any one of the trials can be and has been faulted. They are small, some too brief, and there is significant patient loss in some which was not perfectly accounted for. But in the aggregate thiazide treated patients had about one half the number of new stones, as those untreated, adjusted for the numbers of cases: 22 vs. 45%. For citrate the difference is more marked.
Synergy Between Diet Sodium and Thiazide
Reduced sodium diet alone will lower urine calcium – I have already shown that above. Thiazide lowers urine calcium by increasing proximal
tubule reabsorption of sodium and water, and secondarily of calcium. High sodium diet can partly overcome that effect so urine calcium does not fall as much as it might with less sodium, and, as is well known, potassium will be lost in excess in many people.
The Borghi diet and thiazide trials provide data on urine calcium vs. urine sodium with and without thiazide. I have compiled their data in a spreadsheet for those interested.
In the trials urine calcium fell with  urine sodium in the control arms of the diet and thiazide trials (red and blue circles, respectively). and in the low sodium high calcium arm of the diet trial (blue triangles). But thiazide (black triangles) lie below all other points at corresponding urine sodium levels. This means that thiazide and low sodium diet are synergistic.
urine sodium in the control arms of the diet and thiazide trials (red and blue circles, respectively). and in the low sodium high calcium arm of the diet trial (blue triangles). But thiazide (black triangles) lie below all other points at corresponding urine sodium levels. This means that thiazide and low sodium diet are synergistic.
Effects of Potassium Citrate On Stone Risk Factors
Potassium citrate has the potential to reduce calcium stones by reducing urine calcium and by increasing urine citrate itself which binds calcium in a soluble salt complex and is an inhibitor of crystallization. Therefore one might expect a more complex relationship between stone prevention and the single index of supersaturation.
Trial Data
Unlike the thiazide trials, the citrate trials offer only scattered urine chemistry data that do not lend themselves to the kind of pooled analysis I provided for thiazide. All I can do is consider each one as a separate experience. Each header below links to the PDF of the trial report.
Ettinger trial. Of the three long term trials, only that of Ettinger provided data on urine calcium before and during citrate treatment, and the treatment citrate data do not include corresponding urine sodium data so I could not plot them as I did on the preceding graph. Patients had active recurring stones (>=2 within the prior 5 yr and >=1 in the past 2 yr) and stones were >50% CaOx. Treatment was 63 mEq citrate/d, 21 mEq Mg and 42 mEq K, given in 3 divided daily doses for 3 years. Diet for placebo and drug arms was restricted in salt, sugar, oxalate, and animal protein with no more than 2 servings of dairy product – a low calcium diet.
Mean baseline citrate excretions were 549 and 587 placebo and treated groups, respectively, and corresponding treatment values were 548 and 769 mg/d. Urine SS was not altered for CaOx or CaP nor were there differences between placebo and treated groups. Given that stone rates fell (stone data in stone spreadsheet) without a change in supersaturation, the best possible hypothesis is that the increase of urine citrate increased inhibition of calcium crystallization.
Barcelo trial. Patients had 2 or more stones in 2 years and ‘low’ (less than 2 mmol/d) or low normal (less than 3.4 mmol/d) urinary citrate. Given a MW for citric acid of 192 mg/mmol these are 384 and 652 mg/day as selection criteria. Stones were calcium oxalate, alone or mixed with calcium phosphate – amount not specified. Mean urine citrate in the normals used by the lab was 3.36 mmol/d or 645 mg/day. Mean urine citrate was 347 and 370 mg/d at baseline in the treatment and placebo groups, respectively. With treatment urine citrate rose to 633 mg/day in the active arm but remained at 384 mg/d in the placebo arm. Urine calcium is said to have not fallen – no data presented. Supersaturations were not measured. So in this trial patients were initially low enough in urine citrate to pose risk, and treatment reversed that risk by raising urine citrate appreciably. Stone recurrence indeed fell (stone data in stone spreadsheet).
Hofbauer trial. Patients were ‘idiopathic calcium oxalate’ stone formers. Urine citrate at baseline (mg/d) was 243 and 249 and, at end, 282 and 438 mg/d, placebo and treated groups, respectively. There was ‘a marked decrease in the 24-hr urine calcium…’ but the data are not shown. Citrate dose was individualized to maintain urine pH between 7-7.2 so there was no double blinding. Dietary restrictions were imposed on everyone, but not specified. So the patients had low enough urine citrate to pose a risk of stones, and treatment increased urine citrate and lowered urine calcium but stone recurrence was identical in both groups (the red and blue circles on the trial figure, stone data on the linked spreadsheet). In fact both groups had rather low stone rates at the end, comparable to those of the treated arms of the other two long trials. So although stone risk should have differentially fallen, stone formation did not show a differential response.
Soygur Trial. Patients stone free or not after SWL were given 60 mEq/d potassium citrate in 3 divided doses and new stones or growth of fragments noted at 12 months. Stones were CaOx. Details are in the spreadsheet. About half of the patients had urine citrate excretions below 320 mg/day – their cutpoint for ‘hypocitraturia’. In the 20 citrate treated hypocitraturic patients mean urine citrate rose from 234 to 525 mg/d and from 343 to 718 in the remaining 26 who were not hypocitraturic. So reduced stone crystal formation – as one might put it – associated with increased urine citrate excretion.
Lojanapiwat Trial. Post SWL or PNL 76 patients were followed over 1 year for new stones or growth of stones. Stones were ‘calcium containing’ but I could not find more detail. Of these, 39 received potassium sodium citrate 81 mEq total citrate in three divided doses. Mean urine citrate was 304 vs. 259 mg/d and 305 vs. 405 mg/d, control vs. treated groups, baseline and at one year of treatment, respectively. So reduced stones were associated with an increase of urine citrate.
rSummary of the tials. The Hoffbauer trial poses real problems because urine citrate rose yet stone formation did fall. Better put, stone formation fell alike in treated and untreated patients. In the other trials, stone risk and stone rates fell in tandem. If I had to choose between more thiazide and more citrate trials I would vote for the latter because results are more variable.
Synergy Between Diet Sodium and Potassium Citrate
The trials offer no insight into the effects of potassium citrate on urine calcium, as opposed to urine citrate, and whether a synergy exists between diet sodium and urine calcium. We would expect potassium citrate to lower urine calcium by offsetting diet acid load.
 Among 197 patients selected only as consecutive studies in our kidney stone clinic, 32 had been prescribed potassium citrate (open circles on graphs) the rest no drug treatments. Those given potassium citrate had a higher urine excretion of potassium, higher pH, and lower urine calcium excretion, evidence for use of the agent. This link leads to the complete statistical analysis of these data.
Among 197 patients selected only as consecutive studies in our kidney stone clinic, 32 had been prescribed potassium citrate (open circles on graphs) the rest no drug treatments. Those given potassium citrate had a higher urine excretion of potassium, higher pH, and lower urine calcium excretion, evidence for use of the agent. This link leads to the complete statistical analysis of these data.
Urine calcium varied with urine sodium (Upper left panel). Those people taking potassium citrate (open circles) had significantly lower urine calcium excretion rates at corresponding urine sodium excretions – dashed line (detailed analysis in the linked document).
From the few clinical trials that provided both sodium and calcium excretions with and without potassium citrate treatment (upper right panel) those on the agent had lower urine calcium (open circles, statistics on page 9 of the link) and the lower urine calcium was statistically significant adjusted for the urine sodium. These data come from trials beyond those considered in this article.
The bottom two figures will be of interest only to a few scientists. They show urine calcium vs. the delivery of calcium out of proximal tubule and also delivery as a function of urine sodium, left and right panels, respectively. The analysis is on pages 4 and 5 of the linked document. Potassium citrate reduces urine calcium at any delivery but the p value is borderline (0.058). Potassium citrate reduces distal delivery adjusted for urine sodium (lower right panel of figure, page 4 of linked document) p=0.003. So potassium citrate reduces urine calcium and in part does so by increasing proximal tubule reabsorption.
The clinical message is this: Although data are incomplete and a proper trial needed, it would appear that reduced sodium intake and potassium citrate act independently to reduce urine calcium excretion, so they are synergistic, as are reduced sodium diet and thiazide. But the trials are not sufficient to confirm these as hoc observations.
Observational Data – Urine Supersaturations
Citrate can reduce urine calcium and raise urine citrate which would lower supersaturation, but will also increase urine pH which would increase CaP supersaturation. Once again, the trial data – shown above – are very limited. From our 197  observational points, however, we have some idea of what will happen clinically.
observational points, however, we have some idea of what will happen clinically.
Urine SS CaOx (upper left panel) is significantly reduced by K citrate, but the reduction for SS CaP (upper right panel) is not significant. SS CaOx does not vary significantly with urine sodium excretion with (dashed line, open symbols) or without K citrate treatment (lower left panel). It is much lower with K cit than without (6.63 vs. 3.72, sodium adjusted p<0.001. CaP SS varies strongly with urine sodium (F=6.2, p=0.014) and the sodium adjusted value for SS is lower in K Cit treated subjects (0.73 vs. 1.13, p=0.03). The details of the GLM are at the end of the linked document.
I suspect these shards of observational data will hold up in a prospective randomized trial. Differences are significant with only a few treated subjects, and the distribution of points is reassuring. So for the moment these data can be pressed into service as a guide to treatment pending a formal trial.
A STRATEGY FOR PREVENTION
Here is, so to speak, the payoff for all this reading and analysis. Essentially I offer my own clinical practices, but have offered the reasons my practices are as they are. I have based them on the analyses you just read, and you are free to inspect the data themselves and come to your own conclusions.
Before Treatment
Be Sure Stones are Active
I cannot say this often enough as nothing can prevent old stones passing. One needs to know what is there at the start and that new stones are forming in the kidneys or stones are passing not previously seen in the kidneys.
Be Sure You Know The Stone Crystals
Our purposes are to stop crystals from forming stones and given this purpose what could be more unfortunate than ignorance of those crystals. Old analyses are no guarantee for the present time. Things change. Stone analysis is not very expensive compared to unnecessary surgeries.
Be Sure the 24 Hour Urines Represent Life as Lived
How else to say this but that urine collecting is burdensome and people like to do it while at home whereas they live a majority of life elsewhere, on the go, working, and doing all those things we call real life. Likewise, once treatment begins, collections have to represent what is being done every day, not simply on collection days. When collecting urine, patients must not drink especially well, or follow diet guidelines especially well. Physicians cannot assure that urine samples represent life, but patients can and need to.
Use Fluids and Diet First
Raise Urine Volume above 2.25-2.5 l/d or more
I do not scruple or demur about fluid intake; no amount up to 5 liters a day is too much in an otherwise healthy person. Too much can make life unacceptable, so one compromises. But I try for the most a patient is willing to achieve.
The water trial achieved about 2.6 liters of urine volume, the diet trial about 2.1 liters – but there were other treatment measures. My own studies of supersaturation vs. urine volume point to about 100 ml/hour or about 2.5 liters daily, and the Curhan data place the minimum for absolute safety about 2.25 liters. So water intake to achieve 2.25 – 2.5 liters of urine flow will be perhaps 3 to 3.25 liters in a routine environment. It is a high goal that many will achieve perhaps only some of the time. Hot environments and extra physical activity require more water. Like every physician I remind patients that when their urine looks dark and concentrated it is concentrated, and crystals do not sleep.
What I say here about water and other treatments concerns idiopathic calcium stone formers without systemic causes of stones and without important systemic diseases that would affect such treatments as water loading, low sodium diet, potassium loading, or thiazide diuretics. I mention this here because patients can and should read this article and those with any significant medical concerns must always seek the opinion of their physicians in regard to treatments.
Prescribe The Kidney Stone Diet = US 2015 – 2020 Diet Recommendations
The recent US diet recommendations more or less exactly match the needs of our patients: Intakes are Sodium below 2,300 mg/d, Calcium 1,000 to 2,200 mg/d, protein 0.8 gm/kg/d, refined sugar below 10% of total carbohydrates, and potassium intake 112 mEq/d from fruits and vegetables. Other articles on this site detail the diet and the correspondences between it and the needs for stone prevention. Given vast resources have been spent to vet the science behind the health benefits of this diet for such major diseases as osteoporosis, obesity, diabetes, and hypertension, there is no need to scruple about its use in any idiopathic stone former unless one has some serious additional diseases.
Lower Diet Oxalate
GIven the evidence that 1,200 mg calcium intake markedly reduces urine oxalate, it may prove less arduous in practice to control urine oxalate than with the present much lower calcium intakes. I estimate that diet oxalate intakes between 100 and 200 mg/d will be adequate for most patients. Our oxalate lists are as reliable as we could make them thanks to the efforts of Dr. Ross Holmes and the basic excellence of the Harvard lists we started with. Our article culls out the highest oxalate foods, so pruning the food choices can be easier. The arduous nature of diet change is perhaps lessened by the wide variety of foods still available.
Because oxalate is an issue for stone formers and not for people in general, the need to place high calcium foods into meals that contain appreciable oxalate must be brought to our patients separately from information about the diet in general.
Test the Effects of Diet Change Before Prescribing Drugs
Effects on Subsequent 24 Hour Urine Collections
My approach is to make the changes in hydration and diet, and see what happens to urine stone risk factors – urine supersaturation especially. If things are changing for the better, and there seems some room for more fluids or more diet change, patients are probably well advised to make those changes and measure again so as to achieve the most that they can achieve.
The caveat is that the urine collections be realistic reflections of life as lived. This means that the diet and fluid changes are universal not simply reserved for days of collection. If fluids and diet changes lower supersaturation well below what it was when stones were active, the use of a drug is hard to justify unless patients say they cannot or will not pursue these measures long term or despite lowering of supersaturation stones continue to form. This latter simply means that relevant supersaturations are still too high.
Effects on New Stone Formation
Many patients form no more than a stone every year or so, and if fluids and diet lower supersaturation in multiple samples I am in favor of waiting to see if more stones in fact appear. But this is said with reservations in all dimensions. Patients have their own desires as to level of protection vs. risks and troubles of medications. There are innumerable subtleties – single kidneys, kidney damage, many kidney stones in place, very high rates of stone formation, special risks from stone passage, job or other life situations that make another stone a special problem, pain medication issues that are better served by promptness. Even so, I push back against prescribing drugs at first in hope of the best control as a baseline on which to add medications – sodium, water, sugar, calcium, and oxalate.
In this matter of achieving the best from conservative measures and best timing of medications resides the very soul of medical practice. That is why I cannot put what I do on the page in a simple manner, and why I despise stepwise guidelines that pretend to substitute for real encounter with the complexity of human goals and of life as lived.
When Should Fluids and The Kidney Stone Diet Begin?
It is Best Begun After One Stone
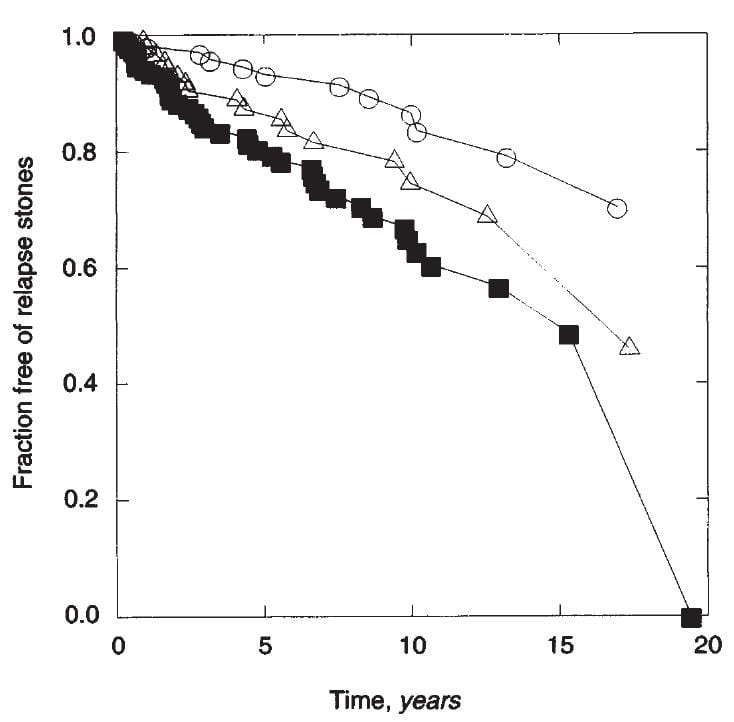 In 1994 I reviewed our experience with the outcomes of treatment in patients – all men, I am afraid, because of limitations of numbers – to the number of stones that patients had formed before treatment was begun.
In 1994 I reviewed our experience with the outcomes of treatment in patients – all men, I am afraid, because of limitations of numbers – to the number of stones that patients had formed before treatment was begun.
Although these are not trial data, merely observations, we had a long time of treatment – along the horizontal axis. Also treatment was never one thing but whatever mixture of fluids, diet, and medications that seemed right – I was taking care of patients not doing research about that care.
The people who had formed only one stone before treatment (open circles) were about 85% stone free at about 10 years 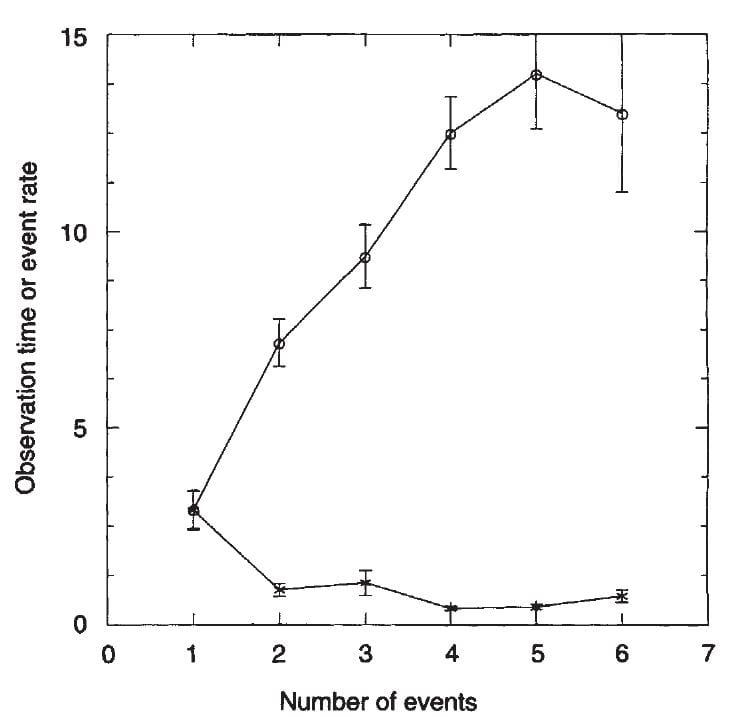 (about 15% had relapsed). Those with 2 stones (as we counted this would have been two clear stone episode periods each of which might have contained more than one stone) were less responsive to treatment, with about 25 – 30% relapse at 10 years. Those with many stones – 3 or more dated episodes and as much as 10 or more stones did least well, about 40% relapsed at 10 years. As this is a life
(about 15% had relapsed). Those with 2 stones (as we counted this would have been two clear stone episode periods each of which might have contained more than one stone) were less responsive to treatment, with about 25 – 30% relapse at 10 years. Those with many stones – 3 or more dated episodes and as much as 10 or more stones did least well, about 40% relapsed at 10 years. As this is a life
table, there are a lot of statistics, and they are in the paper.
Among these same people we could reconstruct the time course of stone formation before we
ever saw them clinically, using our detailed records. The graph to the right plots time of pretreatment observation (gray circles) and rates of stones/year against the number of events.
A rising number of dated stone events was associated with a longer time
between the first stone and entering our program. For example those with one episode came only 2.5 years after the first stone. This graph means the obvious – if you wait more stones may form, and more stones implies a less ideal outcome from stone prevention efforts. That the rate remained flat means the same: Stones formed over time at a more or less constant rate.
This was all observation, so I analysed the trials that I have already summarized for you earlier in this article and asked if the fraction of patients who relapsed despite treatment rose  with a larger number of pretreatment stones. This was not easy because it was not always clear just what was the pretreatment stone average – individual data were impossible to find in most cases.
with a larger number of pretreatment stones. This was not easy because it was not always clear just what was the pretreatment stone average – individual data were impossible to find in most cases.
On the vertical axis I placed the percent of treated patients who relapsed. I had no interest in the placebo cases for this analysis because my question is whether more pretreatment stones prejudice the outcome of treatment efforts. On the horizontal axis is my best estimate of the average number of pretreatment stones in the treated group. I emphasize this is very hard to be sure of, so the graph is an approximation.
Even so the results are remarkable. Relapse despite treatment tends to rise with an increasing number of pretreatment stones. The three points marked ‘Mulit’ are from my paper. I knew the numbers of pretreatment stones exactly. The points at 1 stone are my single stone formers and the single stone formers from the Borghi trial. The others are labeled with the treatments used. The axes are logarithmic because of crowding in the lower ranges,
I cannot use this kind of data to test a scientific theory – too indirect, but I can use it to ask whether the kidney stone diet is worthwhile early on.
It Is Appropriate for Family Members
Stone formation is highly familial. One reason may be the marked tendency for hypercalciuria in the immediate relatives of calcium stone formers which we have documented in children. Because the kidney stone diet is essentially identical to what is recommended for all American people – absent the high fluids and specific attention to oxalate – we certainly should advocate for it in relatives of calcium stone formers.
Add Medication To the Best Fluid and Diet Control Possible
Thiazide
In the absence of other compelling reasons, I add thiazide when stone activity continues despite increased urine volume, reduced urine sodium to below 100 mEq – 2300 mg sodium intake, and I have come to believe I cannot lower relevant supersaturations further with more fluids or less sodium intake or reduction of sugar or animal protein loads. Or, as I just said, when patients are not willing or able to maintain diet and fluid changes.
I strongly avoid adding thiazide when sodium excretion is above 100 mmol/d because it will be less effective and potassium wasting a common problem. I also cannot justify thiazide when urine calcium excretion is already low, below the Curhan lower limit for risk of 200 mg/d.
My own preferences are for indapamide or chlorthalidone as opposed to hydrochlorothiazide. The latter is short acting and has been tried mainly in twice a day dosing which is less convenient for patients. Personally, as opposed to the trials, I begin with 12.5 mg of CTD in most cases and often achieve good lowering of urine calcium and supersaturations. This is because I try to control diet sodium before using the drug.
Potassium Citrate
The US diet calls for 112 mEq of potassium from fruits and vegetables, which is about 60 mEq/d more than present diet averages. For this reason, actual use of the diet should raise both urine potassium – a sign of proper usage, and urine citrate. This latter is because potassium from these food sources will generally be with organic anions that are metabolized in their acid forms. This means a proton is taken up with metabolism, so new bicarbonate will be produced in the blood and such bicarbonate may commonly reduce NaDC1 transporter rates and increase urine citrate.
Potassium citrate pills should therefore be for people with calcium stones and presumably a low enough urine citrate excretion to make such treatment worthwhile – below 400 mg/day despite the kidney stone diet. I understand we have one trial of the agent in people with substantially more urine citrate.
From our observational data, low sodium diet is synergistic in lowering urine calcium and therefore should lower supersaturation. Likewise, although it increases urine pH, potassium citrate lowered urine CaP SS in our observational patient cohorts. Of course, I use this agent for potassium replacement for thiazide hypokalemia. My typical starting dose is 30 mEq/d in three divided doses.
Choosing Between Thiazide and Potassium Citrate
I do not think of this as a matter of taste. Thiazide seems appropriate when urine calcium remains above the Curhan risk limit despite all that can be done with diet, and either supersaturation is not lowered at least by half from baseline, or it has been but new stones are forming. I mention again, because names are confusing, that hypercalciuria used to be defined as against the distribution of values taken from non stone forming control populations. This is fine for physiology research but no longer tenable given the real nature of the urine calcium – stone risk relationship. Therefore the old statement that thiazide is a choice in even ‘normo-calciuric’ patients is misleading; most may have had urine calcium levels above 200 mg/d.
Potassium citrate seems appropriate when urine citrate excretion is below the Curhan risk limit despite diet. Because citrate action is complex, on urine calcium and as an inhibitor of crystallization, a low value seems to call for correction. When urine citrate is already above the risk threshold I am willing to use the agent to correct hypokalemia from thiazide, and usually do so. Because potassium depletion lowers urine citrate, the agent is often appropriate for that reason.
This reasoning means that from time to time I use both agents in an independent manner – for higher urine calcium and for lower urine citrate. No trials have explored their combination.
Calcium phosphate stone formers pose a special theoretical issue. Potassium citrate will increase urine pH and unless sodium is controlled urine pH increase will tend to increase urine CaP SS. No trials have tested this agent for CaP stones per se. I use it but with low diet sodium and attention to whether urine citrate and CaP SS increase and try to make a best judgment.
Allopurinol
I personally do not use this drug in my own work even though I am indeed the person who first put forth the idea that it might prevent stones. In principle, for patients who cannot take thiazide or potassium citrate, have recurrent stones, and all other treatments have proven unsuccessful I would use it at 200 mg.day. In his trial report Ettinger mentioned that it was unlikely the drug would work only when urine uric acid excretion was high, but he had no more reason for this comment than I do in these comments. We have the one trial which is sound, and a treatment that lacks a mechanism and so is difficult to endorse. But although a mere empiric remedy it can have its uses.
What if You Need Help Getting the Diet to Work?
In my practice I find it very difficult indeed, and I know lots of physicians find the same. As for patients and their families, diet is hard to change and even harder to maintain over time. My writing partner, Jill Harris, provides help via the internet, and if you would like to have some here is where to find her: Jill Harris’s Kidney stone diet site
Thank you Dr. Coe for your efforts to understand kidney stone formation and to educate patients and clinicians!
Is there a single article you’d suggest for a patient to show to a clinician to interest them in prevention of Ca Oxalate stones after the first occurrence? The main audience would be specialized Nurse Practitioners and P.A.s because the only time I saw an M.D. was immediately before ureteroscopy to remove a stone of then-unknown composition. The part of the present article from “Here is, so to speak, the payoff” to the end seems a likely candidate for such an article but I fear that is still too long for a clinician to want to read it. (I’m not as concerned about the part after the heading “Add Medication To the Best Fluid and Diet Control Possible” because that is a “next steps” at which point the clinician presumably is already interested.)
On the other hand, your website seems to provide more than enough information for the patient that only a lab for 24h urinalyses (and the cash to pay for them) is needed, not clinician interest. Having myself so far failed to get a clinician to order such urinalysis, I wonder if you have any tips or resources for a patient engaging a lab directly to get this done–or is it really as simple and straightforward as one might hope? To properly gauge the effect of a trial dietary change, should that change be made a day (or more?) before the start of the 24h collection period? Can the analysis be done with a small fraction of the collected 24h of urine (which would make sending via USPS/UPS/FedEx more practical)–e.g. collect 2.4L, send .2L and multiply result by 12 to get mg/day–or must the entire amount be submitted?
Thanks again.
Sorry to submit a 2nd comment already, but I’ve just been looking at LabCorp’s website and Quest (questhealth.com) website, and neither seem to offer a lab test that includes a calcium or oxalate (do I want calcium oxalate supersaturation? Or “free” oxalate?) analysis. So if you or any of your patients or website visitors know where/how to get these analyses, I’d like to know. Perhaps that is among your web pages already and I overlooked it, I don’t know.
Hi Kevin, it is Litholink, wholly owned by LabCorp. Regards, Fred
Hi Kevin, You do not so much have a real problem but what amounts to a roadblock. Why in the world would anyone want to alter diet or use meds to prevent more stones and not get 24 hour urine testing to figure out what was wrong and see if treatments really worked? I would find a better type of physician. Ordering Litholink, the best vendor (I have no financial interest in the company even though I founded it and sold it to LabCorp) requires a physician in most states. Any family physician can do this. And you are right, this site offers a lot of help interpreting the results. But really one should have a physician, which is why I said what I said. As for writing a more forceful article focusing on why 24 hour urines, Jill Harris did a good job. Regards, Fred Coe
I have what seems to be idiopathic hypercalciuria. On a 24-hr urine test, all my levels are within range, except for the calcium which is high. The urinary oxalate is often extremely low-to-undetectable. Should I even be bothering with a low oxalate diet?
Hi Jason, I gather you form stones, and suspect they are calcium based. Given what sounds like idiopathic hypercalciuria oxalate would not be a prime treatment target but rather the high calcium that has many treatments available. I am sorry for the lateness of my reply which arose from technical issues on the site. Regards, Fred Coe
Dr. Coe, thank you again for all the information you have shared with this site. It’s the best resource on kidney stones available to me as a patient.
I hope I can ask you for an opinion.
I was offered Alopurinol and/or thiazide diuretic. Should I take one or the other or both?
Here’s the history.
55 years old male. Passed one 2 mm Ca-Ox stone 4 years ago.
I was told to drink more and take 2 potassium citrate tablets per day (1 g each), no 24h test was done.
2 years later one 3 mm stone was removed by ureteroscopy and laser, there’s 3 mm stone in the other kidney, as seen on CT. Benign tumor was resected from the bladder at the same time.
Since the last stone I had a few 24h urine tests and no more stones. I cannot test supersaturations in Canada.
Looks like I have idiopathic hypercalciuria. By reading your site, I figured I need to limit the sodium.
I drink more, try to limit animal protein and sugar, eat more vegetables and fruit.
The highest Ca I had was 375 with Na at 2800, the lowest Ca was 236 with Na under 1600 (that lab cannot test lower levels). Another test was Ca 284 and Na 1771 mg.
I understand you recommend bringing Ca under 200? Should I take diuretic?
Alopurinol was suggested, because my Ph ranges 5.5-6.5, uric acid 4.5-8.6 mmol, urea 680, 753.
My oxalate is not bad at 19-29 mg and citrate was at 700-900 mg.
I continue taking K Citrate twice a day.
Phosphorous is at 60, 69.
Thank you very much.
Hi LK, You are doing well with reduced diet sodium. Higher meat intake does not associate with kidney stones. Potassium citrate has several positive trials, and you seem to be free of new stones. I would just leave things as they are. If more stones form chlorthalidone or indapamide might be considered by your physicians. When uric acid stone risk arises from low pH potassium citrate is the proper prevention, allopurinol has no important role. Regards, Fred Coe
Thank you, Dr. Coe!
I am confused about Allopurinol. CUA and AUA recommend its use:
“In calcium stone formers, allopurinol is effective in reducing stone recurrence in patients with hyperuricemia but does not provide any benefit in patients with normal serum uric acid levels “
Hi LK, I started this long ago. High urine uric acid excretion was more common in calcium stone formers than normals, I thought, and used allopurinol as a treatment. It seemed to work. My work led to a formal trial which was positive. But we never came to understand how the uric acid fostered calcium stones so I never made much use of my own research. It may well be effective and of particular use when other causes are absent or when treatment options are few. What I really understand is that I need to write a proper article about this for the site. Thanks, Fred
Dr. Coe,
I have three small (<5mm stones) that have been stable x2yrs by CT in my right kidney. These were first noted in 2021. They were not present in 2009. In 2018 I started a high fat low carb physician supervised diet to reduce risk of diabetes. In the last two years, I have also had intermittent bowel obstruction, and have finally had a segment removed. In this time my diet was mainly HFLC, but leading in the six months up to my surgery. I had stopped eating any significant greens or fiber due to abd pain. I was on 12.5 mg of HCT for presumed idiopathic hypercalciuria with Ca 24 of 306 (baseline 345). My serum potassium reached the lower limit during this time and pre-surgically I discontinued HCT to avoid electrolyte issues. Extenuating circumstances are atonic urinary bladder. My bladder does not tolerate supplements such as potassium citrate or B12. In addition, the additional urine volume results in catheterization up to 10 times daily, which has its own increased risk and difficulties. Upping my thiazide would cause me even more issues of urine volume and potassium deficits. I could try a new baseline now that I can eat more vegetables while I stay in LCHF or I could revert to a standard American diet. Do you have any thoughts?
Dr Doe,
I am 46 F.
Worrying about what could have caused high Urine Calcium of 267 (expected<250)
Here are blood work results
All blood work related WBC/diff and RBC are normal
CMP Result as follows:
Urea Nitrogen-10
Creatine-068
EGFR: 109
Sodium:138
CO2:24
Calcium:8.8
VIT D:59
Protein: 6.7
Albumin:4.1
Blobulin: 2.6
Billurobin:2.3
ALP total: 58
ALP Bones Specific : 6.5 ( range 4-20)
AST and ALT both at :12
PTH, INTACT : 29
TSH: 2.05
B12: 627
ANGIOTENSIN: 13
ANA: NEG
I take abt 1000-1200 Calcium intake daily, + multivitamin that 200 calcium daily.
I also take 2000UI Vit D daily.
Hi Tina Roy, I suspect you have genetic (‘idiopathic’) hypercalciuria. Serum values are normal, urine calcium is high, bone disease is a common complication as is stone disease. But one needs a lot more in the 24 hour urine than just calcium. I would recommend a full 24 hour urine stone panel. I presume you have formed stones or have bone disease. Regards, Fred Coe
Thanks Dr Coe. First of all, apologies for spelling your last name incorrect. Also, i tried different approach and stopped multi-vitamin supplement and Vit D supplement for 1 day and reduced my yoghurt diet by 2 spoons only and went to CA 24 hrs Urine again. It came down to 206 by just stopping Vit D , multivitamin and not taking 2 spoons of yoghurt. any suggestions?
Hi Tina, I am not sure about your approach. I presume you have formed kidney stones. I suspect you have idiopathic hypercalciuria. If you are indeed a stone former and have IH then your bone health and stone risk are better served by a reduced sodium high calcium diet. Low calcium diet is not a good thing. As I do not know the real details of your situation this is just a comment but you might mention it to your physicians. Regards, Fred Coe
Hello Dr. Coe,
My wife suffers from kidney stones and they always seem to be infected. She has had 3 ER room situations in the last 3 years and we just received the analysis of the last one. It is 50% Calcium Oxalate Dihydrate (Weddellite) and 50% Carbonate Apatite (Dahllite). She’s 39 years old and otherwise extremely healthy. We are at a loss as to what to do. The urologist we go to is just suggesting taking a look at them every 6 months but that doesn’t seem to be working. Any suggestion would be greatly appreciated.
Hi Jeremy. Here is my best on first steps. I would recommend looking it over and see if it fits in with what you both might want to do. Her stone is 1/2 calcium phosphate so keep that in mind as you read. They are harder to prevent but prevention is certainly a reasonable expectation. Regards, Fred Coe
I am on Hiprex and vitamin C for persistent UTI, are there any supplements/diet I should avoid or add to prevent kidney stones. Thank you!
Hi Julie, diet is not a treatment for infection. As for kidney stones, here is my best effort as an introduction to prevention. Evaluation is the first thing to do so you know what causes the stones. Possibly your stones are arising from infection. Here is an article on that subject. Regards, Fred Coe
Dear Dr. Coe,
I have been advised not to ingest dairy due to cardiovascular issues. I am also lactose intolerant. I know you generally prefer calcium food to supplements but would it be okay to meet the 1,200 mg/day calcium recommendation with Calcium Citrate supplements taken with meals instead? What is the downside to using a supplement rather than food for calcium?
Hi Gwen
With meals, such supplements should be safe. Not much data, I am afraid. Best, Fred Coe
Thank you so much for the great resources on this site!
I’m a 35 year old male (5’10, 150 lbs) and unfortunately experienced passing a kidney stone for the first time 3 months ago. Unfortunately, the CT scan taken at the time showed I have 5 more stones in my left kidney, and 7 more on the right. The stones about evenly distributed between 1mm and 5mm. The stone I did pass was Calcium Oxalate.
I just got back my first Litholink test, and the results were a little disappointing, since I was hoping for something more “actionable”, as I’m highly motivated to figure out what is going wrong in my body. I did my best to be totally representative during the test, since I knew it wouldn’t be useful otherwise. The only change is I stopped taking Turmeric supplements (3g/day) since experiencing the stone, and I don’t plan on resuming them, since I’ve read they can be high in oxalate, so the results should be representative.
Urine Volume: 1900 ml
SS CaOx: 2.12
Urine Calcium: 56 mg/day
Urine Oxalate: 37 mg/day
Urine Citrate: 637 mg/day
SS CaP: 0.37
Urine pH: 6.288
SS Uric Acid: 0.35
Urine Uric Acid: 0.59 g/day
Urine Sodium: 124 mmol/day
My physician flagged my urine volume as slightly low, and my pH as slightly high, but everything else seemed fine, and he was kind of at a loss to provide any root cause. After reading your site I will request another 24 hour test to confirm. He has never even mentioned a blood serum test, but I will bring it up with him as well.
I’m happy to permanently increase urine volume, and permanently avoid the worst of the oxalate foods, spinach, almonds, etc. However, the values make me wonder if something else could be at play? I guess since my SS CaOx is above 1, it could be reduced further, even though it is on the low-side already? Appreciate any thoughts!
Turmeric contains considerable amounts of oxalate: https://www.perplexity.ai/search/amouont-of-oxalate-in-3-grams-kRFtPL.XQaOH3fzZVKJPeg#0. Three gm/d would add about 60 mg and given you have 37 mg/d ambient this could put you as high as 50 – 70 mg depending on efficiency of absorption. From an additional search https://www.perplexity.ai/search/amouont-of-oxalate-in-3-grams-kRFtPL.XQaOH3fzZVKJPeg#1 I get about 9% will be absorbed so probably the actual urine delivery will be much less – around 6 mg/d more, but that can depend on the person and other foods eaten. Also a supplement might generate a lot of oxalate delivery over a short time creating transient crystallization. Given the otherwise normal results, I would guess – pure guess here – it was the turmeric. BEst, Fred Coe