
![]() Some of us overdo things with shakes and powders, some with 2 pound steaks. Others love sweets too much and don’t eat much protein. Like all the diet factors in stone and bone disease, protein intake is complex. Certainly, we all need protein in our diet but how much? Experts debate the best course, and patients wonder what to do.
Some of us overdo things with shakes and powders, some with 2 pound steaks. Others love sweets too much and don’t eat much protein. Like all the diet factors in stone and bone disease, protein intake is complex. Certainly, we all need protein in our diet but how much? Experts debate the best course, and patients wonder what to do.
Abraham van Beijeren was, by the way, little recognized in his day but now considered a major painter of ‘luxuries’ like this standing roast. I chose it, as opposed to others more brilliant, because it looks modern – I have seen something like it on my own dining room table.
In preparing this article I have made considerable use of the analyses performed by professor Tanis Fenton. She graciously read and edited the article up to the details of renal physiology, and the work much benefitted from her expertise which I gratefully acknowledge here. All errors are entirely mine, however, should you find any.
How Much Protein Do We Need?
The National Research Council (US) Recommended Dietary Allowances Tenth Edition (1989) suggests (Table 6.4) 0.8 gm/kg of dietary protein for adult men and women.
A subsequent WHO meta analysis of mostly the same underlying data supplemented by more recent studies comes to much the same conclusions, but in perhaps a more nuanced manner. A more recent analytical critique of the whole matter is not remarkably far off in estimates 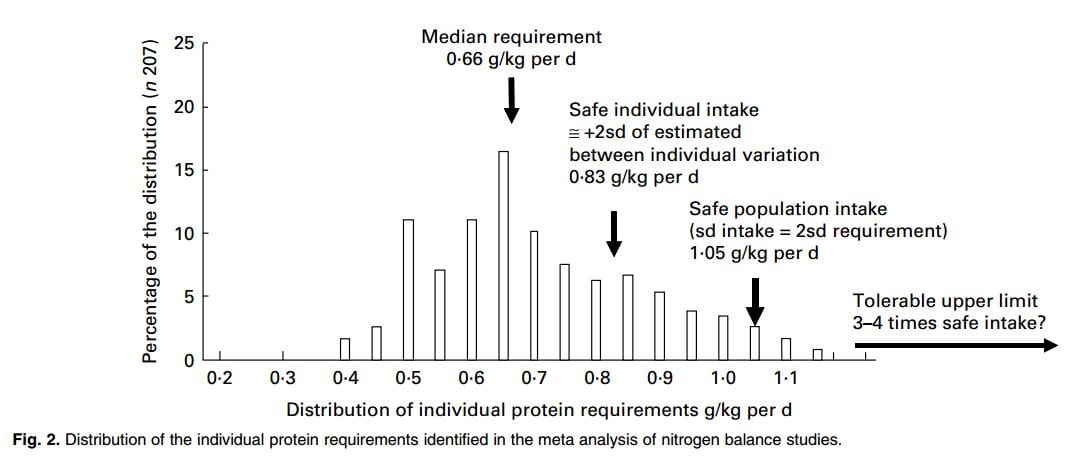 for adults, though pregnancy and childhood seem controversial.
for adults, though pregnancy and childhood seem controversial.
This summary graph from the critique gives a sense of how the protein requirements are set. The median requirement is where about half of all studied subjects were in neutral nitrogen balance – their body protein mass would be stable, a very important matter. The safe population intake is set higher. The safe individual value is high enough enough that 97.5% of the individuals in a population would be in balance: almost all people would not lose protein mass consuming this amount of protein – for example muscle. The ‘Safe population intake’ is set higher. Although the safe individual intake is correct, within a population individual requirements vary, so the recommended level needs to be increased so that 97.5% of the individuals in a population offered that recommendation will be in balance.
That number from the WHO meta-analysis, the safe population intake, is about 1.05 gm/kg body weight/day.
Given the safe individual intake is 0.83 and the upper bound of the intake range is 1.05 gm/kg/d no one should choose a protein intake below 0.83 gm/kg/d and very few need more than 1.05 gm/kg/d unless challenged with unusually high demands for physical work. This means that in helping patients choose a protein intake for stone prevention we work in that range 0.83 – 1.05 mg/kg/d.
The Story in Brief
Because commercial vendors provide a measure, albeit indirect, of protein intake in every 24 hour urine – using the amount of urine urea to calculate the protein catabolic rate (PCR) in gm protein/kg body weight/d – patients know their intake and can control it by diet. This measure is applicable to healthy people who are neither gaining or losing protein mass.
Like sugar, protein loads raise urine calcium, and urine calcium is a major risk factor for stone formation. Low protein intake may reduce urine calcium but is bad for overall health. Whether or not high protein intake raise urine calcium at the expense of bone integrity is fiercely debated right now. Low protein intake is not good for bone.
So with respect to protein intake stone treatment steers between too high and too low.
I could just tell remind you that the high and low markers for normal people – not otherwise compelled to high or low protein diets – are between 0.8 and 1 gm/kg/d and be done, but that is not my way. I believe understanding is the key to long term treatment and encourage patients to read about protein. Physicians already know everything I write about, but many way enjoy a ride through a familiar and charming countryside.
Protein Imposes an Acid Load
For several generations we have known that the sulfur containing amino acids cystine and methionine produce 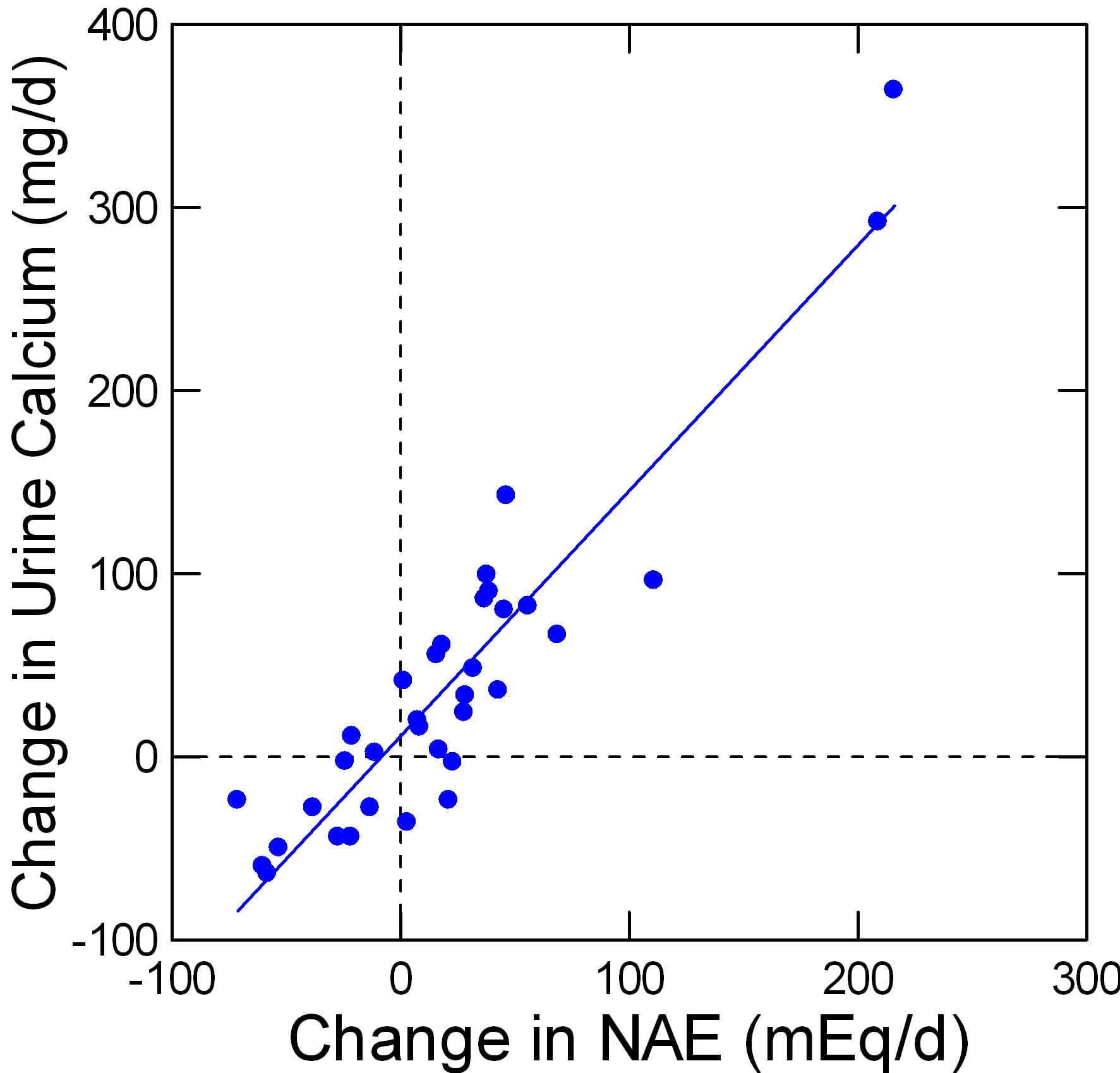 an acid load and that rising diet protein acid loads correlate with increased urine calcium excretion. Giving acid loads experimentally increases urine calcium excretion.
an acid load and that rising diet protein acid loads correlate with increased urine calcium excretion. Giving acid loads experimentally increases urine calcium excretion.
Some believe acid loads promote bone fractures by mobilizing bone mineral stores and that alkali treatments prevent this form of bone loss. Others believe that the protein increases urine calcium by increasing intestinal calcium absorption and does not adversely affect bone,
Fenton and her colleagues performed what I think is a rigorous ‘meta-analysis’ of studies available up to 2006 concerning effects of acid load on the urine calcium excretion. The acid loads were varied by variation of diet protein, by giving alkali such as potassium citrate and by giving acid loads like ammonium chloride – a purely experimental strategy. No matter how acid load was varied urine calcium varied linearly.
The points on this graph are from Table 2 of her paper. I have redrawn her figure to suit my taste. My data set is available for others.
The negative changes in net acid load are from alkali loading such as potassium citrate. The changes from, as an example, 50 mEq of alkali – the -50 position on the horizontal axis – corresponds to a fall of about 50 mg/d of urine calcium.
In my article on prevention of calcium stones, we found the effect of potassium citrate corrected for urine sodium was in fact -92 mg/d, which is very close to the results from this large analysis (see page 8 of the statistical analysis linked from the article).
Acid Loads Apart From Protein Raise Urine Calcium
I would be remiss to leave matters as if Fenton’s meta-analysis were a sufficient guide to 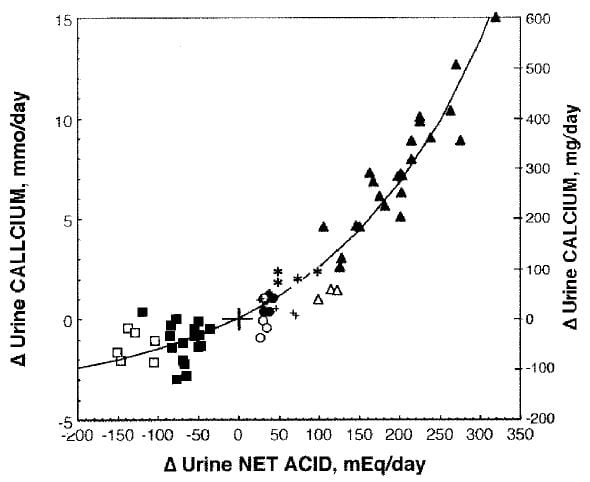 this vast literature. Her approach emphasizes the quality of the human trials. Another review more emphasizes the underlying technical problems of assessing net acid base balance. In this work acid base balance was altered by NH4Cl (closed triangles), methionine (open triangles), egg white (*), beef (closed circles), soy protein (open circles), deprived of KHCO3 (+), given KHCO3 (closed squares), or given NaHCO3 by replacing some of the dietary NaCl and maintaining Na intake constant (open squares).
this vast literature. Her approach emphasizes the quality of the human trials. Another review more emphasizes the underlying technical problems of assessing net acid base balance. In this work acid base balance was altered by NH4Cl (closed triangles), methionine (open triangles), egg white (*), beef (closed circles), soy protein (open circles), deprived of KHCO3 (+), given KHCO3 (closed squares), or given NaHCO3 by replacing some of the dietary NaCl and maintaining Na intake constant (open squares).
Despite the differing formalisms and even scientific instincts of the investigators who reviewed the topic (the one a group of skilled analysts, the other a group of expert acid base physiologists) the overall result is amazingly uniform. For example at about -50 net acid excretion, there would be about 50 mg less urine calcium and at about 200 mEq of extra acid about 300 mg more urine calcium in both studies alike.
Whereas the Fenton points easily fit a linear regression, the larger range of the Lemann review shows the response is not linear but has a curving character. If you look closely at the Fenton points there is indeed a slight sag around 0 meaning that perhaps a curving regression might have a higher multiple R2.
The point of showing all this is obvious: However you review the papers, acid loads increase and alkali loads reduce urine calcium, meaning this is a vigorous phenomenon, not some houseplant that cannot stand up to the weather. It has been found in many laboratories over many decades, in humans – shown in these two figures – and animals alike.
Does Protein Itself Raise Urine Calcium?
By this one must mean does a change in diet protein raise urine calcium more than would a corresponding increase in net acid load from some other source. Alternatively, it could mean what happens if one gives a protein load with enough alkali to offset the acid from the protein.
Four studies appear to fit the needs for data in that they are trials of protein loading in a rather 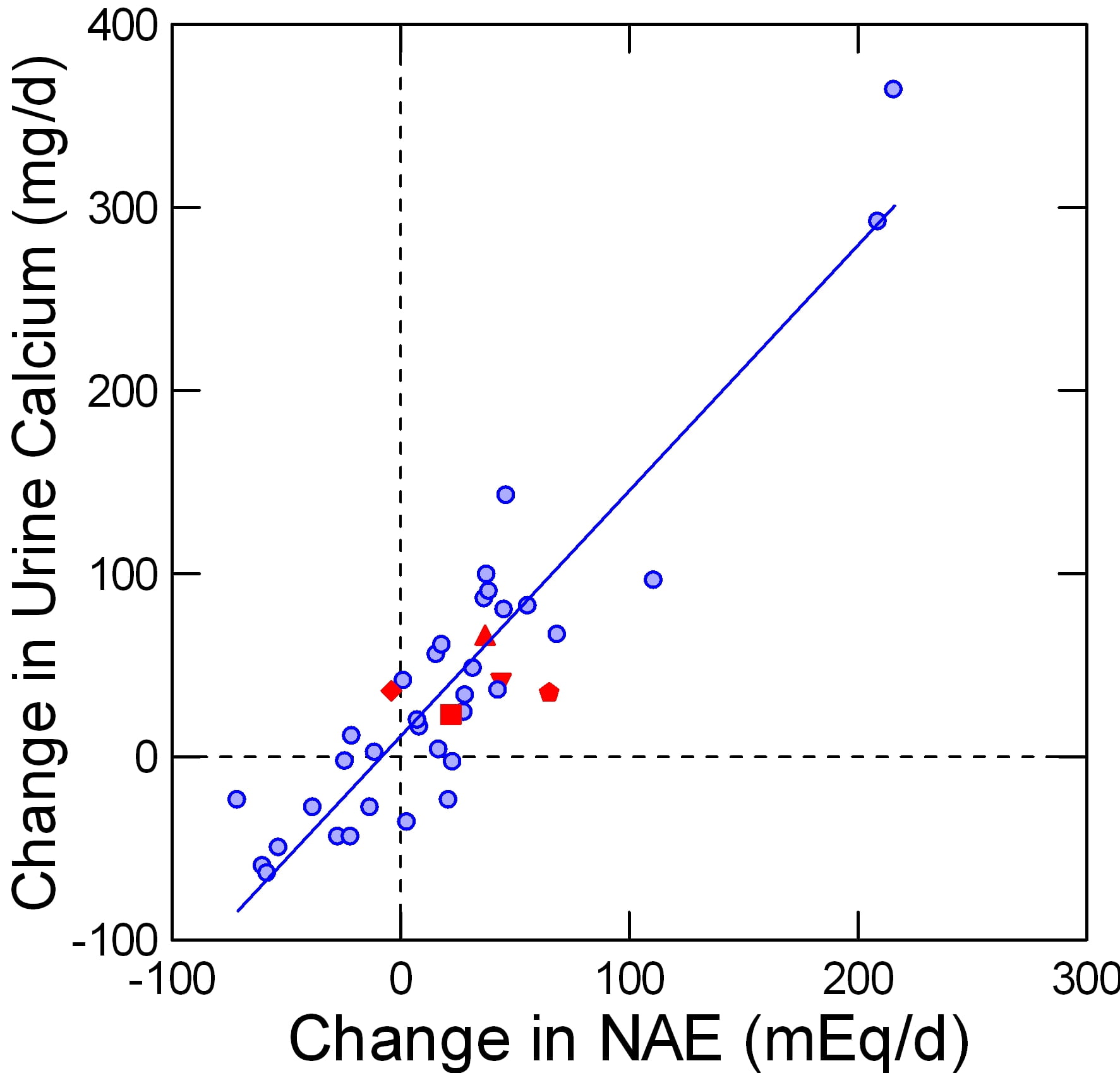 pure form, using foods and with considerable care for total nutrition. Of these, one used alkali to offset the protein acid load. The points taken from these additional reports are here.
pure form, using foods and with considerable care for total nutrition. Of these, one used alkali to offset the protein acid load. The points taken from these additional reports are here.
The Fenton data are in a faded blue, for visual contrast. The protein load studies are in red. The pentagon and diamond are protein load +KCl and the same protein load + K Citrate in a single trial.
In these trials, protein intake was varied over a two fold range, mostly from about 1 to about 2 gm/kg/day.
More or less the data fall on the Fenton plot.
In the one special trial it is obvious that the K Citrate (diamond) lowered the change in NAE without affecting the change in urine calcium. So it is possible to dissociate a protein effect from its acid base effect within the controlled environment of a trial. Given the modest quantitative changes in NAE it is possible that natural variability of urine calcium excretion might have permitted apparent stability despite the lack of a change in NAE in one point and a significant increase of NAE in the other, but the statistical testing is based on the observed variability and gave a low probability from chance alone.
Essentially the trial of diamonds and pentagons tells us that protein itself has a renal effect on calcium handling.
Does Diet Acid Load Damage Bone?
I am not so unwise – being an expert in stone disease not bone disease – to enter into this debate on my own. All I can do is present recent reviews by the real experts who do not seem to have convinced one another.
Meta analyses up to 2011
Fenton has performed what appears to be a rigorous screening of available balance studies 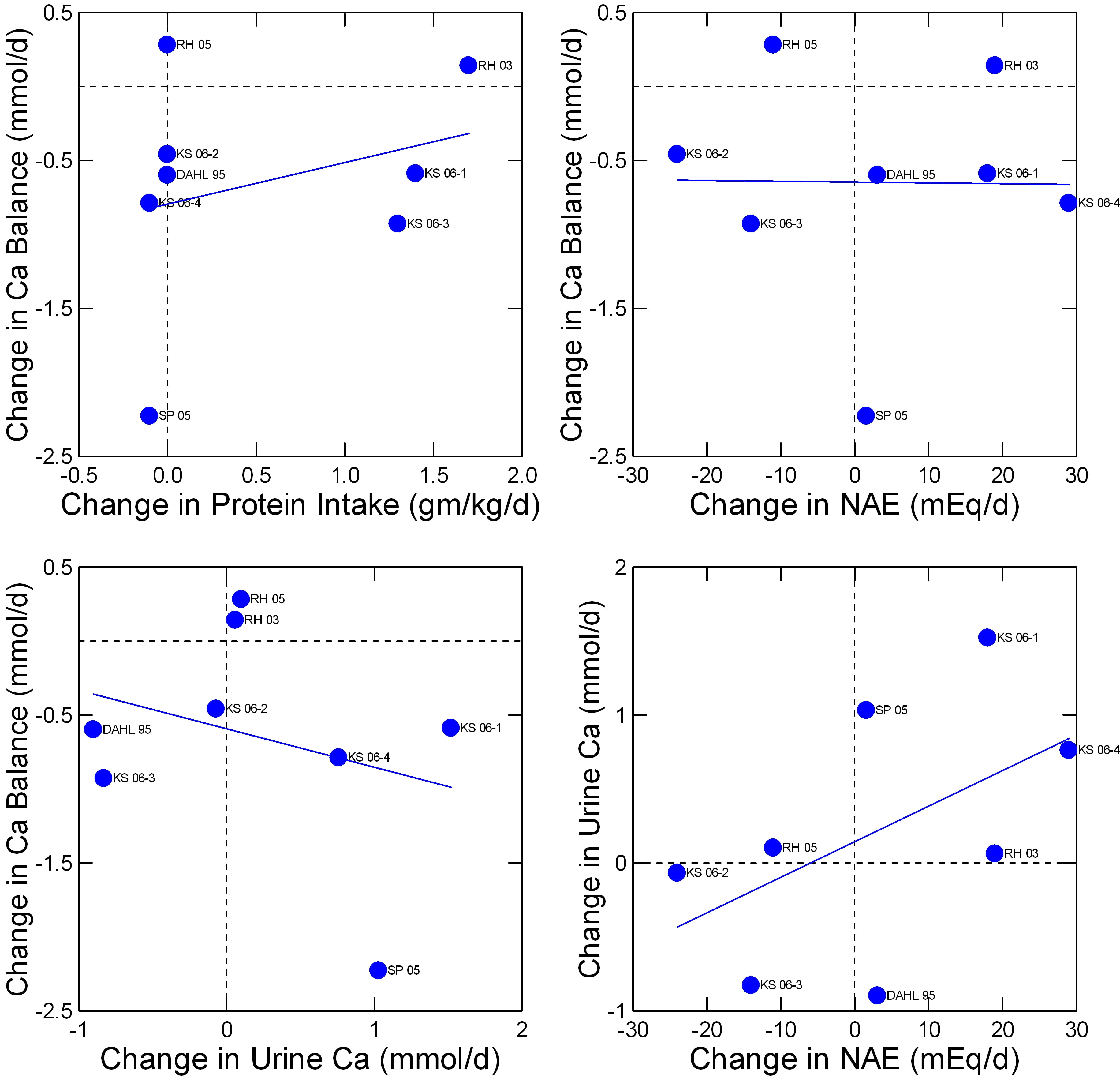 concerning the effects of acid loads and protein intake. She culled out eight studies that met the criteria used by the Institute of Medicine in their assessment of dietary requirements for calcium and vitamin D. Her dataset is not presently available to me but I have replotted her summary data in a manner I find ideal for this site.
concerning the effects of acid loads and protein intake. She culled out eight studies that met the criteria used by the Institute of Medicine in their assessment of dietary requirements for calcium and vitamin D. Her dataset is not presently available to me but I have replotted her summary data in a manner I find ideal for this site.
The abbreviated names refer to her Table 2 in the reference. The data I extracted and plotted are here.
Change in calcium balance does not vary importantly with change in protein intake (upper left panel) nor at all with change in net acid excretion (NAE) from the protein amounts and types (upper right panel). There is perhaps a slight inverse relationship between change in balance and change in urine calcium (lower left panel) and, as in less curated studies shown above, a marked direct relationship between urine calcium and NAE (lower right panel).
The only way balance (upper right panel) can be indifferent to change in NAE and yet inverse on urine calcium which is itself dependent on NAE is that changes of intestinal calcium absorption make up the difference from calcium lost in the urine. One presumes this but proof may be beyond the resolution of calcium absorption measurements.
Balance Data after 2011
At least one important research group performed what appeared to be a fine balance study in which potassium citrate was given to neutralize diet acid load and calcium balance was measured by an expert in such work. The data for urine 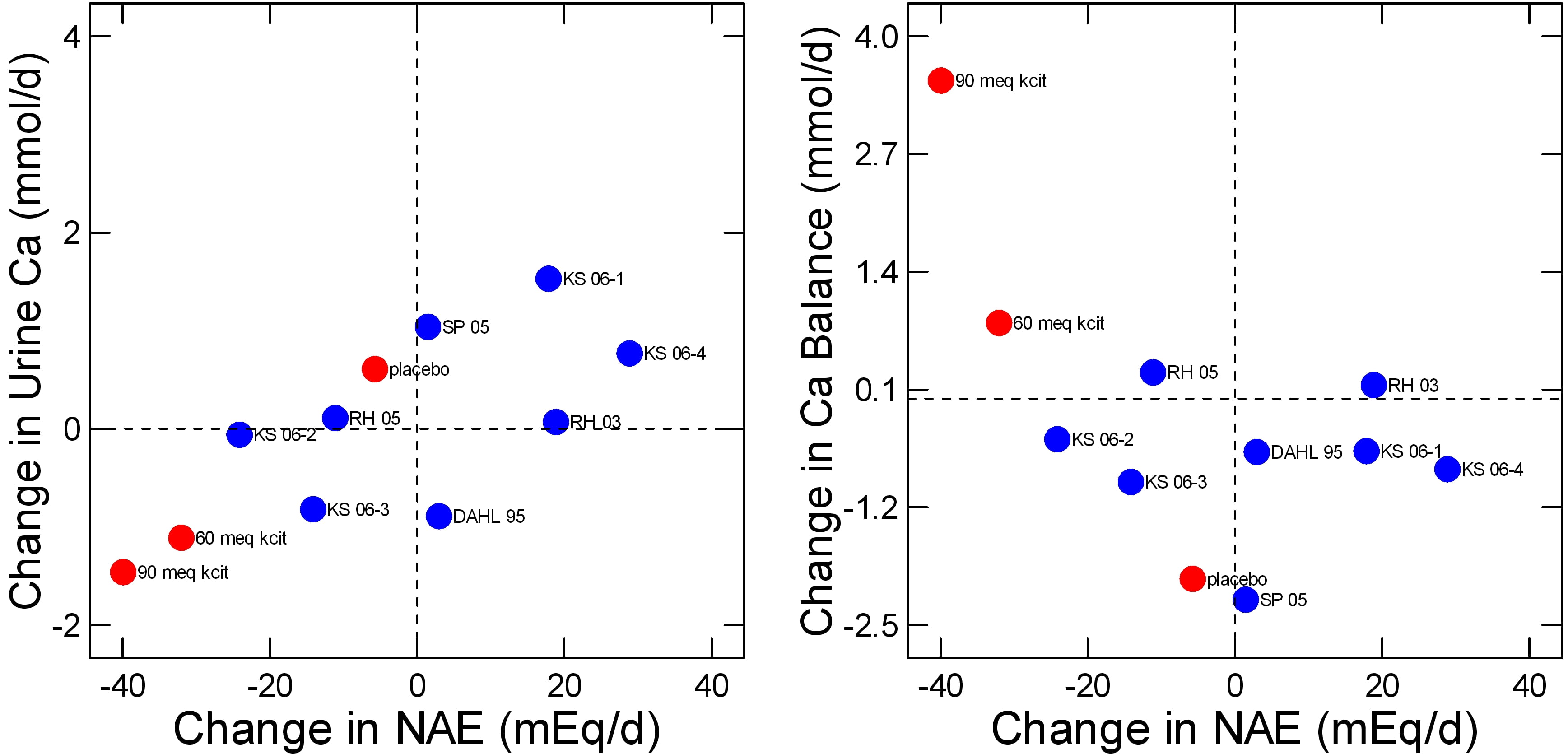 calcium and balance are in mg/day and I simply transformed them to mmol/d for graphing purposes.
calcium and balance are in mg/day and I simply transformed them to mmol/d for graphing purposes.
The way the new study was performed was to compare change in NAE and both urine calcium (left panel) or calcium balance (right panel) after 6 months of treatment with placebo, or with 60 or 90 mEq/d of potassium citrate. I have plotted the new study points in red over the Fenton analysis in blue and have removed linear smoothers for clarity.
In a sense all of the studies agree about urine calcium. A change in NAE gives rise to a reasonably stable and predictable change in urine calcium here as in the much larger data sets I already reviewed.
For balance (right panel), the placebo and 60 mEq potassium citrate points are not out of range of the other studies Fenton considered although with these two points – and of course their many more internal measurements – there might be a slight inverse relationship between NAE and balance. The single 90 mEq point would make a more powerful negative regression, as is obvious.
Whether or not the Fenton group would consider this study as being of the highest quality I do not know, as I am not a student of meta analysis or of the rather elaborate and – to me at least almost incomprehensible – criteria for ‘high quality’ as opposed to ‘low quality’ studies. But if this study were indeed part of the elect minority it might change opinions.
Perhaps I should say something, however, and it is this. Potassium citrate does lower urine calcium just as acid loads raise it, and so far as I can tell the agent will not worsen bone mineral balances. So in giving it I have no reservations about bone just as I cannot have definitive hopes for better bones as a result, at least right now.
Sodium Intake Affects Urine Calcium Response to Acid
In a prior article I showed the urine calcium lowering effects of potassium citrate were independent of the effects of diet sodium. But those data, and trial data like it are observations. Suppose you give people an acid load so their urine calcium 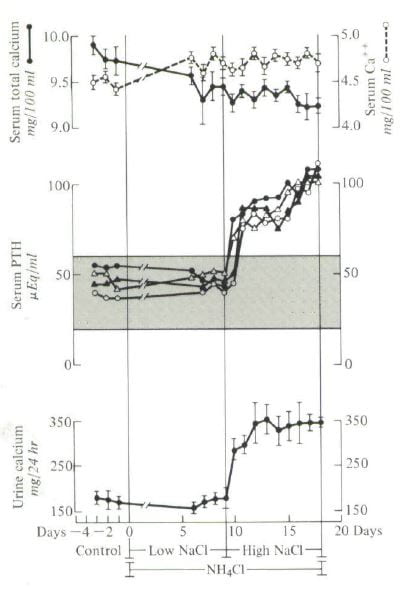 goes up as a result of what you did and also varied sodium intake – a direct experiment, not observations.
goes up as a result of what you did and also varied sodium intake – a direct experiment, not observations.
I did that, and although others also may have done the same, my experiment was a good one and I like it.
Four people were studied during three control days – points to the left on the graph. They had normal urine calcium excretions (lower panel), normal serum PTH levels (middle panel) and normal serum total calcium (upper panel filled circles) and ionized calcium (open circles). That is what one expects from normal people.
I gave them ammonium chloride which is an acid load, and as you might expect by now their urine calcium should go up. But, I also lowered their sodium intake to 40 – 80 mEq daily (average about 1500 mg) and – take a look – urine calcium did not change.
Their blood became distinctly acidic – you will have to look at the paper. The calcium ion rose and the total calcium fell because the acidity tends to liberate calcium ion from binding to blood proteins and from phosphate complexes.
I then raised the diet sodium to about 200 mEq (4000 mg) and there was the urine calcium increase. Serum PTH went up, too, perhaps because the kidneys were losing calcium.
All of this experimental data emphasize the importance of diet sodium. Given our protein intake imposes a tonic acid load, low sodium diet should presumably act as it did here and lower the calcium losses acid loading may well create. In a well performed study of bone mineral balance in menopausal women, it was the combination of reduced sodium intake and increased calcium intake that brought bone mineral balance into a positive range.
Given this, I would add to my prior statement that low sodium diet is itself safe for otherwise normal people and is likely to benefit bone health, or at least do no harm to bones.
How Does Acid Base Change Alter Renal Calcium Handling?
A Fair Warning
What comes is technical.
Physicians and scientists know this material and can read it fluently. If you are not one of those, you can read what is coming and find is very interesting. I avoid jargon, and explain things reasonably well.
However response to acid or alkali loads is a fundamental property of kidneys, crucial for life, deeply embedded in the way they function and elaborated and refined by millions of years of evolutionary biology. It is, therefore, elegant in its way and hard to grasp.
Overall Kidney
Many investigators have shown that chronic acid administration raises urine calcium by reducing tubule calcium reabsorption. My data are as good as any to illustrate the  point.
point.
Here are the same four people from the graph just above. The vertical axis shows urine calcium excretion and the x axis shows the measured filtered load of calcium, the rate at which calcium is being filtered out of blood into the fluid moving down the tubules of the kidneys.
At any filtered load, urine calcium was lowest when people ate a low sodium diet without an acid load (closed circles). High sodium diet without acid raised urine calcium, as expected (filled triangles). Chronic acid load with low sodium diet lowered filtered load (open triangles are to the left of the filled triangles) but urine calcium was slightly higher.
High sodium and acid load caused very high urine calcium excretion at any filtered load – reduced tubule calcium reabsorption.
Let me put this into perspective. In other articles about treatment I have shown you much the same thing. Sodium raises urine calcium and that is why low sodium diet is a good treatment for stones. Moreover, the effect of sodium on urine calcium is far more marked in stone formers with idiopathic hypercalciuria than in normal people. What I show here is that acid loading makes normal people behave like those with IH – urine calcium becomes overly sodium dependent. The high sodium diet raises the urine calcium far more than it could without the without the acid load – the sodium load + acid load points are far above the other points.
Proximal tubule
We have already taken trips down the nephron. For those who have forgotten the road, here is a good review. The nephron tour begins at about page 3 of the link.
Likewise here is a good review of what is to come concerning how kidneys respond to acid loads. Read it later.
What I have Said Already
In the oxalate review noted just above we focused on SLC26a6, which can permit chloride and oxalate or other negatively charged species to move through the cell membrane facing the tubule fluid. In another article we considered the citrate transporter that faces the same way and reclaims filtered citrate. That transporter is regulated by acid and alkali and I presented the outlines of that regulation.
As part of the citrate article I introduced NHE3, a sodium hydrogen exchanger that moves protons into the tubule fluid, reabsorbing filtered bicarbonate, and also moves ammonium ion (NH4+) out into the lumen.
Chronic acid loading increases NHE3 and therefore proton transport and therefore the completeness of bicarbonate reabsorption. This latter means less alkali (bicarbonate) can be lost in the urine – and is instead returned to the blood to counteract the acid load. Also less is present in later parts of the nephron. This means that protons secreted into the tubule fluid downstream are not used up reabsorbing bicarbonate but can flow into the urine as acid excretion.
The article also told us that acid loads increase the conversion of glutamine, an amino acid, to α-Ketogluterate which process takes up protons thus generating alkali. The extra ammonia from the nitrogens of glutamine is moved into the tubule fluid by NHE3 and migrates downstream. Eventually much of it leaves in the urine. We say the sum of the protons titrating phosphate and the ammonia in the urine are the net acid excretion (NAE), which we spoke of in relation to protein acid loads in this article. The ammonia counts as acid lost because metabolism of the α -Ketogluterate produces new bicarbonate.
What about Sodium?
As NHE3 moves protons into the tubule fluid to titrate bicarbonate and therefore reabsorb most of what was filtered, it reabsorbs a sodium ion for each proton secreted (therefore called a sodium hydrogen exchanger). That sodium is transported out of the cells at the blood side of the cell by the ancient and universal sodium potassium active transporter (ATPase) and reenters the blood. In fact one might point out that NHE3 runs off the sodium current created by the ATPase.
NHE3 is like a revolving door with people coming in and coming out but those coming in are doing all the pushing. This is because the sodium concentration in the cell is kept very low by the ATPase so there is a driving force favoring sodium entry into the cells from the tubule fluid.
In the process of reabsorbing sodium, the proximal tubule reabsorbs bicarbonate via NHE3. This reabsorption of bicarbonate makes the concentration of bicarbonate in the tubule fluid lower than that in the blood. The proximal tubule cells are linked together by junctions that are rather water permeable, so as sodium and bicarbonate are reabsorbed water will move with its dissolved salts back into the blood. There is more to this, but perhaps I can add it later.
Effects of Acidosis on Sodium Reabsorption
This should be simple. More NHE3 from acid means more sodium reabsorption and more bicarbonate reabsorption, therefore more water reabsorption. So proximal tubule sodium reabsorption will go up. In the proximal tubule, calcium is reabsorbed almost completely passively – meaning along with sodium. Therefore with acid loading calcium reabsorption will also go up.
But that is not what happens.
Chronic metabolic acidosis in rats produced by ammonium chloride administration, reduces sodium reabsorption in both the proximal tubule and along the nephron downstream from the proximal tubule. There is no increase in filtration of sodium but rather a reduction of tubule reabsorption. This means that calcium reabsorption would necessarily be reduced. In fact, I just showed you in my own experiment how acid loads reduce calcium reabsorption.
How can it be that NHE3 can increase and yet sodium – and therefore calcium – reabsorption go down?
This problem led to an important experiment in animals.The rats were given acid loads and as expected bicarbonate reabsorption increased significantly. But, there was trouble with respect to chloride – an element we have not as yet mentioned.
Sodium reabsorption via NHE3 is limited by the fact that only 20% or less of the sodium filtered uses bicarbonate as its counter ion – negative ion to balance the positive charge on sodium. The rest – a majority – uses chloride, and I did not say how chloride is reabsorbed. It is in fact reabsorbed passively in part because of the sodium bicarbonate reabsorption; as bicarbonate is reabsorbed with sodium and water more and more of the sodium left behind is with chloride so the concentration of chloride rises compared to blood. This chemical concentration difference causes chloride to move back into the blood, but through what can it pass?
It moves through chloride exchangers – more revolving doors, and one of these is our old friend SLC26a6, the oxalate transporter.
 What if that transporter is not working right?
What if that transporter is not working right?
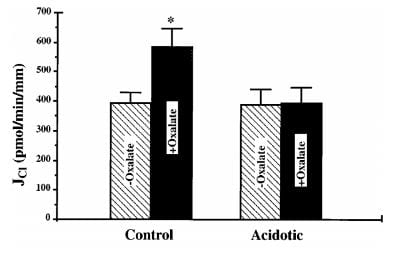
You would find what was found in these rats. Sodium bicarbonate reabsorption was above normal when the rats were loaded with acid but chloride oxalate exchange was not functioning. This means that sodium chloride reabsorption could not proceed at a normal rate so neither could sodium chloride and water reabsorption – or calcium reabsorption.
In this bar graph, the control rat kidney cell membrane transport of chloride (vertical axis) rose markedly when oxalate was added – black bars so labeled. Membranes from the acid loaded rats lost their oxalate response. Another pathway, chloride formate exchange, was also abolished.
When sodium reabsorption is thus limited, calcium reabsorption is also limited, and therefore the stage is set to deliver more calcium downstream as without acidosis, and potentially more out into the urine.
This oxalate requiring transporter is SLC2a6, our friend from oxalate days. One must wonder if acid base changes affect renal oxalate handling.
Distal Convoluted Tubule (DCT)
I am skipping over the thin and thick ascending limbs of the loop of Henle. Though dear to me, there is a powerful effect of acid in the DCT that can greatly affect urine calcium excretion and this article can be only so long
We have not much spoken about the cells here, but I will tell you that they have a protein in them that can ferry calcium through the cell from the urine side to the blood side and a transporter to move calcium out of the cells into the blood.
Most importantly they have a gated transporter that controls entry of calcium into the cells, bearing the stately name TRPV5.
When the outsides of 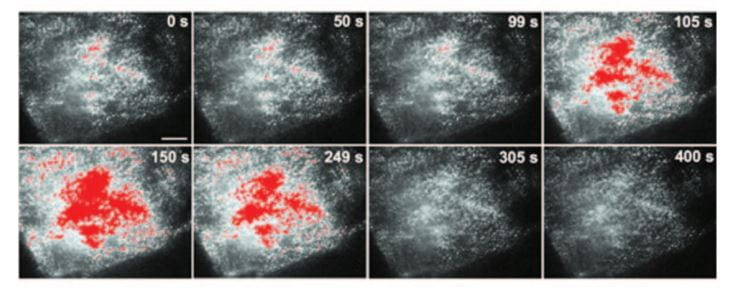 the cells are made alkaline, more of these transporters are moved into the cell membranes facing into the tubule fluid.
the cells are made alkaline, more of these transporters are moved into the cell membranes facing into the tubule fluid.
Transporters moved from just inside cell membranes into the membranes themselves show up here as the colored splotches in these images of single cells. The whole experiment is over 400 seconds. The pH of the medium bathing the cells was raised, and then lowered again. During the high pH period the transporter entered the membranes and could therefore increase calcium entry.
Chronic acid loading lowers the pH in blood slightly and lowers the pH in the fluid delivered to the DCT – because bicarbonate reabsorption in proximal tubule is more complete. This in turn will reduce membrane TRPV5 and therefore less calcium can enter the cells and move back into the blood.
The transporter can be put into a larger host cell than its native DCT cell so its function can be studied in isolation. When this is done, one can demonstrate that the pH outside the cell controls the entry of calcium.
In this experiment the transporter is anchored in the membrane, so the effects on calcium transport are related to the intrinsic carrying capacity of the transporter itself. As the pH is lowered (pHe, x axis) the calcium entry falls from 100% (at pH 9) to nearly 0. The solid line labeled ‘WT’ shows results for the normal transporter (wild type) whereas E522Q refers to a transporter genetically modified so as to alter the pH receptor at amino acid 522.
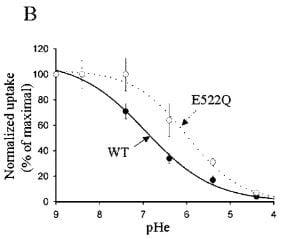 The pH range of interest is of course not from 9 to 4, but from about 8 to 4.5 which is the range over which the tubule fluid pH might vary. When alkali loads are
The pH range of interest is of course not from 9 to 4, but from about 8 to 4.5 which is the range over which the tubule fluid pH might vary. When alkali loads are
given, potassium citrate as an example, the tubule fluid pH can easily be as high as 7 or more because protons are added downstream from DCT in the collecting ducts. This could permit alkali loads to increase calcium reabsorption markedly without necessarily a corresponding increase in urine pH. WIth acid loads the fluid reaching the DCT would be more acid than normal because proximal tubule bicarbonate reabsorption is more complete via the increase in NHE3 that acid loads produce.
TRPV5 is regulated by many factors other than acid base change and therefore urine calcium itself is multiply regulated. Here is an excellent review that lists some of the important regulators. In other articles I will explore these additional regulators.
Synthesis – Clinical Recommendations
Protein is more or less the acid load to which most of us are exposed lifelong who eat a Western diet. All carnivores and omnivores like ourselves have adapted to this kind of acid load over vast expanses of evolutionary time, and the systems we have that are affected by acid loads and can respond to acid loads include the kidneys, no doubt the GI tract – that can secrete bicarbonate, for example – and bone. It is partly because diet protein imposes an acid load that potassium citrate is a useful medication. It neutralizes that acid load and in so doing lowers urine calcium losses and potentially raises urine citrate losses. Given in amounts above net acid load, it imposes an alkali load that can in principle lower urine calcium further.
As for diet protein, the correct amount is certainly that recommended by WHO: between 0.8 and 1 gm/kg body weight/day. Very high intakes are not unpopular, and if pursued will raise urine calcium and stone risk. For various reasons, some may wish or need to use such diets. Probably potassium alkali will reduce urine calcium to some extent by offsetting the acid load. The one experiment that employed protein loading and potassium alkali, however, showed that urine calcium rose anyway, so alkali cannot be relied upon to compensate for high protein diet.
Sodium and acid interact as if partly independent of each other, in that acid load raises urine calcium at any level of sodium excretion presumably by varying filtration and proximal tubule reabsorption via pathways separate from chloride exchange. That makes low sodium diet and alkali a powerful synergistic approach to stone prevention. Possibly low sodium diet will reduce the increase of urine calcium from high protein diet; this has not been tested.
Although protein can raise urine calcium, it is unclear if spontaneous variation of diet protein raises stone risk on average. In his male cohort studies, Curhan found at most a weak and variable association between diet protein intake and stone onset. There have been no convincing trials showing that low protein diet reduces stone production.
Whether alkali spares bone mineral loss from diet protein is a vexed issue because the range of acid load from food is modest and balance changes might be lost in the general variability of the measurement. One cannot therefore recommend chronic alkali for bone health or condemn meat eating as a sure path to bone disease. With time perhaps this important matter will become clearer. But, there is no obvious benefit to high sodium intakes and high sodium intake certainly did interfere with the achievement of bone mineral uptake in at least one well done study, so the use of reduced sodium diet – 65 to 100 mEq (1500 to 2300 mg) daily – is prudent and should be a national health measure.
Way too technical for me to understand. What is the bottom line and how much protein a day should I consume at 230 lbs. in ounces not metric.
Hi Vince, so for a 70 kg man – about 140 pounds – we are talking 70 grams of protein; meat is about 20% protein cooked weight that would be 350 gms or 0.35 kilogram; there are 2.2 pounds in a KG so this would be 0.8 pounds cooked meat. For you at 230 pounds that would be 1.2 pounds of cooked meat a day. That is a lot! Maybe I should put this into the article. Thanks, Fred Coe
This is very helpful. Other sources have conflicted, and produced confusion.
Some sources do not make clear that grams protein do not exactly equal grams meat. Also, the cooked weight vs. raw weight issue is often neglected.
So, for my 100kg self, my low target would be 400g cooked meat/ day? 400g=.88lb=14oz cooked!
That seems quite liberal, have I got this right?
(My stones are primarily 80% uric and 20% Ca oxylate, though some small ones are pure uric.)
Again, sincere thanks!
MikeB
It is 0.8 gm/kg/day protein at the low end and with various assumptions about protein/oz meat your number is reasonable. Uric acid stones are treated with potassium citrate, and that will offset whatever you eat. Be sure and get 24 hour urine testing if you alter your diet to be sure urine pH is above 6. Regards, Fred Coe
Hi sir,
Just wanted to ask if the protein intake is for a normal adult (basically, leading a sedentary/slightly active lifestyle).
If so, what about the students who are training with weights, like 5 days a week. Can they safely go over 1.06 gm/kg/day ? If yes, what’s the safe threshold.
Hi Mike, for stone formers, It makes no difference, high protein raises urine calcium a lot – or can – and therefore stone risk. Safe for stone risk is 1 gm/kg/day. Regards, Fred Coe
Hi Dr. Coe,
Thank you for interpreting the source articles to make this complex topic more accessible to patients. I found this article particularly interesting because we’ve found that protein, and pH, in addition to sodium, seem to have a very large impact on my numbers.
This might be of interest to you…After reading Dr. Orsen Moe’s research, I’ve tried inexpensive genetic testing (intended for researching ancestry) because their raw data include many SNPs reported to be related to incomplete dRTA or acid excretion. I did not find any noteworthy deviations.
My nephrologist suspects that my innate high pH 24 (around 7 before any K citrate, and meat/poultry intake within the new recommendations) may simply be a result of having relatively alkaline blood. She has found no evidence of any disease, just powerful IH. I have no known lung issues.
Can you possibly suggest any mechanisms behind innate high pH for urine, and probably blood, that might fit this puzzle? Appreciate all your help.
Best regards, Al
Hi AL, The best course for you is to lower sodium intake as far as possible – I believe urine pH will fall. I also suspect you might have some underlying genetic disorder but that is hard to guess about from so far away. Regards, Fred
Hi Dr. Coe,
This is really puzzling. The data seems to show this going in the opposite direction:
I’ve cut my salt my more than half consistently for months and have a new SS.
(Note: Infection and kidney disease have been ruled out. Did add Chlorthalidone, and protein is down, but the striking difference is salt):
Before: Na24=195, PCR=1.5, pH24=6.771 (no meds, serum K=4.1)
After: Na24=86, PCR=1.2, pH24=7.121 (12.5 mg Chlorthalidone, high K diet but no supplements, serum K=3.8)
Does this offer any better clues for possible mechanism behind my innately high pH?
Protein seems to be the dominant factor in my SSs, but my doctors have run out of ideas. If something hits you later, please consider dropping me an e-mail. Thanks so much for all your invaluable help!
Al
Hi Al, a high potassium diet will always provide organic anions like citrate – which raise urine pH but also raise urine citrate. Such a diet will lower urine calcium and usually SS with respect to CaP will fall. I do not see these here. So what you have is fine, and I am not concerned unless CaP SS is higher and your stones have CaP in them. Regards, Fred Coe
Hi Dr. Coe,
Appreciate your help very much. Unfortunately, that is exactly my problem. My stones are 80/20 CaOx/Cap (apatite), and although Ca24 has decreased a bit, both key SSs have risen as has pH.
I have reduced salt by >50%, reduced meat by >30%, reduced refined sugar and oxalate intake, added Chrlothalidone, raised volume, and kept serum K up as much as possible to maintain Cit24 as you taught me.
Yet these tried and true dietary changes, even the sodium reduction, seem to be doing more harm than good.
My doctors and I are stymied. Increasing the same seems unlikely to suddenly reverse the trend. Infection and kidney diseases have been ruled out, and I’ve never had SWL. 25 mg Chlorthalidone alone didn’t raise pH, but K Citrate (40 mEq a year ago) raised it to 7.5 so we stopped the K. But pH has never gone back down below 7, possibly due to progressive dietary changes. I just don’t know.
Here is the most comparable pair of full results for looking at SSa as an example. Perhaps you can think of something we missed? Best regards and have a wonderful Thanksgiving,
Al
Last year, before treatment: (Ca intake ~1200mg/day. No meds. Serum K=4.0. )
Na24=173, K24=108, Mg24=237, P24=0.827, NH424=30, Cl24=203, Sul24=50, UUN24=14.50, PCR=1.6
Vol24=3.41, SS-CaOx=3.36, Ca24=378, Ox24=30, Cit24=549, SS-Cap=1.47, pH=6.685, SS-UA=0.12, UA24=0.729
Latest: (Ca intake ~1000mg/day. Salt reduced consistently for months. 12.5 mg Chlorthalidone, high K diet but no supplements. Serum K=3.8.)
Na24=86, K24=80, Mg24=199, P24=0.863, NH424=53, Cl24=115, Sul24=38, UUN24=10.65, PCR=1.2
Vol24=3.69, SS-CaOx=4.04, Ca24=323, Ox24=38, Cit24=597, SS-Cap=1.88, pH=7.121, SS-UA=0.04, UA24=0.829
Linearly adjusted to estimate for Vol24=2.5 as a sanity check: SS-CaOx=5.96, SS-Cap=2.78. Not good!
Hi Al, Things are very complex in the stone field once you get into the atomistic particulars of any one patient. You are indeed a very sharp observer and see this. Because I practice the art of stone prevention as a consulting physician I see detailed issues like yours in almost every patient. But I cannot venture into these details for any one person on this site as it is in the area of public education, not medical practice. If your physicians and you want me to come to grips with what is really happening at this detailed a level, with iterative measurements I am afraid I would be practicing medicine in relation to your care and that requires a more formal kind of relationship with sharing of all data and a purposeful reporting to physicians of record. This is not to be unhelpful, it is just that we are at the point where I would be going beyond an educational role into details that could specifically affect care. Regards, Fred Coe
Hi Dr. Coe,
Of course, that is more than reasonable. Sorry for putting you on the spot!
I find your education through this site, and your personal help applying it to be invaluable. Really appreciate all you and your associates do.
Best regards,
Al
hello doctor thank for this great article, Please help me, my urologists doctors have recommended me to eat meat (any type) only 3 times a week .. that way my protein levels are very low and I am very very worried, should supplement the animal protein to acquire 0.9 gm/kg body weight day? what should I do? I do weight exercises 3 times a week and I have more than 15 stones in the kidney and none have left, next month I will be evaluated to know if they will have to remove them with letrotricia or laparoscopy since I have a cortical cyst of 1.5 Centimeters in a kidney, what do I do with animal protein? since I can not eat the vegetable by the oxalates
Hi Francisco, as the article points out meat is not much of a cause of stones, and low protein diet has no special place for their prevention. Animal protein is in particular no special risk. You need a proper evaluation to find the cause of your stones, and here is a good starting place to read in. Don’t do treatments until you know what you need to treat. Regards, Fred Coe
Love your website! My Humana health insurance newsletter “Silver Sneakers” just sent info saying “To maintain muscle mass and proper functioning, older adults need to eat double the amount of protein that they needed in their younger years.” so I looked online and Mayo, NIH, and TodaysDietician+++ websites seem to agree–new research seems since 2014. Mayo says, “Newer research is showing that higher levels are needed for adults age 65 and older. Between 1 and 1.2 g/kg a day seems to be the target for healthy adults. Those with sarcopenia may need 1.2 to 1.5 g/kg a day.” Others go as high as 1.5 g/kg a day. Is that consistent with what you were saying? Should those of us who are seniors bump up the protein–even if we are calcium stone prone? (You told me earlier that I am a perfect person for the low oxalate diet which I am doing!) Weighing in at about 70 kg (155 lbs), times 1.2 g/kg a day, that is about 84 grams of protein right? Which equals how many lbs of cooked meat??? And does the target protein number include all the protein in the milk products we are eating or just the cooked meat????
Hi Marty, the value of 1 – 1.2 gm/kg/day is all protein and certainly a fine range to be in. Higher amounts of protein will raise urine calcium, but probably not cause bone disease as the article says. I cannot tell you how much meat gives now much protein because meats vary and cooking too. But the US diet guidelines have lots of such information. Regards, Fred Coe
Dr. Coe,
I’m working with a personal trainer to build muscle and lose a little more weight (I’ve lost some weight already from the kidney stone diet). He is recommending a macro nutrient balance of 30/40/30 fat/carbohydrates/protein during the training period, which for my caloric intake, is 146g of protein. Once I’m done, I can take less in to maintain. Based on the WHO recommendations for my weight, I should be taking in no more than 99g per day.
My questions are these:
– Does the type of protein matter relative to stone production risks, or is all protein created equal? I get most of my protein from dairy versus meat and plant-based sources (though moderate amounts, because of oxalate).
– Are there ways to mitigate the added protein, such as additional sodium reduction? (I admit that I got a little cross-eyed when reading the above analysis.) I’m very good about staying under 1500mg of sodium a day, but could further reduce it would provide further offsets.
Thank you,
Mike
Hi Mike, This kind of protein intake will raise urine calcium, as noted in the article. If short term, alright, but not for years. Dairy protein is ot a stone risk, so is preferable, as is plant protein. Regards, Fred Coe
Hello Dr Coe,
I have had some calcium oxalate stones (100% weddelite) removed by endoscopic lithotripsy. Urine analysis revealed severe hypocitraturia and hyperoxaluria and I am now focussing on prevention of new stones via the dietary recommendations on your site, in combination with taking daily potassium citrate supplements (for the hypocitraturia). My blood analysis was pretty fine, except a high concentration of bicarbonate due to low intravascular volume (not necessarily too much bicarbonate). As far as I understand, this kind of reduced oncotic pressure is also increasing the risk for kidney stone formation. I am drinking nearly 3L liquid (mainly water) a day. My doctor told me it could have to do something with a lack of proteins and he recommended me to eat many eggs (though only egg white, i.e. egg yolk removed). I wondered whether you could point any mechanism according to which these recommendations would make any sense. Is a lack of dietary intake of albumin a risk factor for kidney stone formation? Any help or reference to publications would be appreciated. Thanks in advance for your help. Kind regards, Wim.
Hi Wim, The potassium citrate will increase serum CO2 content – bicarbonate – so volume contraction need not be invoked unless there is some other factor you have not mentioned. I do not understand the phrase ‘reduced oncotic pressure’, that would be rare in someone with normal kidneys. Protein feeding likewise has no obvious role concerning serum acid base parameters, so I cannot really understand why one would be on this particular path. Given normal kidneys, the kidney stone diet – protein at 0.8 gm/kg/day, alias the ideal US diet, is fine. If you have a reduced serum albumin, that means inadequate nutrition, something indeed to fix, but a normal diet should be enough absent bowel disease. Regards, Fred Coe
Thank you for your invaluable efforts with MSK and kidney disease in general. I hope you enjoy this article and its resources. God bless. https://nutritionfacts.org/2018/02/08/the-effect-of-animal-protein-on-the-kidneys/
Hi Tomas, Sorry my fonting lacks the a superscript. As you can see from the article you posted on, the case for meat causing stones is very weak, so weak one can say the effect is only with very excess amounts. Kidney disease is another matter, and vexed, but also outside the scope of this site. Diet protein has been a raging debate since at least 1920 with little final solution. Probably the best answer right now is at the low end of the normal range – 0.8 gm/kg/d, but real data proving this matters is hard to come by. As for red meat, everyone blames it, but when you look at the data they are scant. For example, protein loads cause acid loading, but eating a steak is much less of one because it contains significant amounts of metabolizable anions. I cannot write about all this here, as I said. Regards, Fred Coe
I’m more than happy to uncover this website.
I wanted to thank you for ones time for this particularly wonderful read!!
I definitely loved every bit of it and I have you saved to fav to
check out new stuff in your blog.
Hello Sir! I had a stone last year (June 2020) and underwent medical therapy (diuretics, potassium citrate). It was gone last February 2021. I am currently going through weight loss program which requires me to take 1.2-1.7 gm/kg/day for strength training. How long can I follow this diet? I am planning to go back to 1.0 gm/kg/day once I am done with my program.
Hi Vincent, I do not know how long is safe. Let’s say it raises urine calcium and stone risk so keep up the highest urine volume practical and end the high protein when you can. Regards, Fred Coe
Hello Dr.Coe , It was certainly a pleasure reading this detailed analysis about the role of protein in kidney stone formation.I did want to ask though , will there be any difference in the amount of urine calcium(and hence the kidney stone risk) if I consume protein from vegetarian sources , say milk , pulses , soy and so on? I reckon not , since protein is protein at the end of the day , but would love to hear any insights you might have.
Hi Sam, Vegetable protein has no effects to increase stone risk, and neither does animal protein or dairy protein. The only exception is very high animal protein (above 1 gm/kg/d is the beginning of high risk and the available data do not permit finer distinctions; I suggest 1.5 gm/kg/d is too high). Milk proteins seem to lower stone risk. Regards, Fred Coe
Dr. Coe,
Just clarifying, does vegetable protein or dairy protein still raise urine calcium even though there is no stone risk from either? I was assuming any protein would still show some increase in urine calcium.
Hi GF, No – these proteins do not raise urine calcium or stone risk. Fred
Hello Dr Coe, thank you for your wealth of knowledge in this area. It has truly been helpful.
I was hoping you could clarify this point a little further: If I were eating animal protein at the recommended levels (about 0.8g/per kg/day) and then had vegetable protein on top that increased my total daily diet protein intake above 1g/per kg/day (to a level of 1.2g protein/per kg/day, for example), would this raise my stone risk? Is it only animal protein that needs to be kept within the 0.8g-1g/per kg/day range?
I ask because weight training has been a lifelong hobby of mine and ACSM guidelines for building muscle suggest at least 1.2-1.5g/per kg/day of protein intake, but I’d rather not do something that would increase stone risk. So, if I could offset animal protein with vegetable protein (with a pea protein powder, for example) to get me to my ideal protein intake per day I’d do that. However, I am worried that doing so would put me down a path of increased risk. For this question assume that I’m keeping diet sodium & calcium intake at ideal levels as well.
Again, thank you so much for your help on these issues!
Link to ACSM guidelines I cited above: https://www.acsm.org/docs/default-source/files-for-resource-library/protein-intake-for-optimal-muscle-maintenance.pdf
Hi Alex, I do not think so. Protein raises urine calcium generally. But perhaps plant protein is less effective. Here is my suggestion. Use your formula and get a 24 hour urine and see if urine calcium is indeed high enough to raise stone risk – and higher than without the higher protein. That would be a good investment of time and money. Regards, Fred Coe
Hello Dr. Coe, thanks for your helpful advice!
Put more simply from my comment above – do you think I would be generally safe at keeping my diet protein at about 1.2g/kg per day? This would be from all protein sources (animal, plants/oats, & dairy) which for my weight is about 88g/day. Assume my diet calcium, sodium, and water intake are at or near ideal levels.
I’m finding the extra protein (above 1g/kg) helps me stay satiated better throughout the day from my active lifestyle. Thanks again!!
Hi Alex, 1.2 gm/kg/day is not unreasonable. Fred
Hello Dr. Coe, I see meat, dairy, and vegetable proteins were discussed in this article, but what about protein from grains like oatmeal or rice? Do they raise urine calcium/stone risk at all? Thank you!
Hi Jim,
No they will not.
j
Thanks for sharing informative article, I really like this post.
Greetings, Doctor. I am a 63 year old male who drinks protein shakes after a workout. I have had both bladder and kidney stones and my urologist has recommended not drinking protein shakes every day. What say you?
Hi Mike, High protein will raise urine calcium and can promote stones via higher calcium salt saturations. So I agree with your physician. Fred