
 Primary hyperparathyroidism (PHPT) is a systemic disease caused by an excess of parathyroid hormone secretion. It causes calcium kidney stones but also multiple other abnormalities, especially of bone. Here, I am concerned with that subset of PHPT patients with kidney stones.
Primary hyperparathyroidism (PHPT) is a systemic disease caused by an excess of parathyroid hormone secretion. It causes calcium kidney stones but also multiple other abnormalities, especially of bone. Here, I am concerned with that subset of PHPT patients with kidney stones.
A Curable Cause of Kidney Stones
Unlike most stone formers those with PHPT have a good chance at permanent cure. This makes detection of PHPT a paramount aim for patients and their physicians. As I shall tell you, PHPT raises serum calcium above normal and is detected from blood test results. The elevation of serum calcium can be slight and variable so patience and persistence matter a lot. Once diagnosed, PHPT can be cured by a surgery that modern instruments and techniques have made very safe and highly successful in the hands of expert parathyroid surgeons. That is my story.
A Remarkable Experiment of Nature
As I shall attempt to tell you, the strict, fierce regulation of the parathyroid, bone, kidney, intestinal mineral axis normally admits of little variation of the serum calcium level despite rather large traffics of calcium and phosphate in and out of the body, and in and out of bone every day. Dozens of well known and lesser known hormones and signalling pathways, castellated in almost innumerable looping feedback relationships, maintain in serum an amazing calmness and steadiness.
But imagine if one, just one of the hormones involved, and not any one but one among the prime hormones, the kingly olympion hormone goes awry. Resets itself as indeed a royal monarch, a pharaoh might suddenly choose to change the direction of power. No scientist could contrive such an experiment in humans except perhaps for a few hours or days. But nature can do this, does do this, and allows us to observe what happens everywhere in the immense net of controllers when one, of its own, chooses to set the requirements of the body aside and pursue its own treasonous direction.
You might wonder at the picture: The Pharaoh’s Hosts Engulfed in the Red Sea, Lucas Cranach the Elder,1530. To Pharaoh the Hebrews’ desertion seemed treason, to Moses the behavior of Pharaoh a violation of an ancient pact. The very sea itself seemed an outrage of nature, to the Hebrews, their salvation an uncanny thing. To me, Chanach seems an invariable treat to look upon, a miracle painter. Most of all, the sheer mass of it, the vastness of details – is this not what I shall be writing about?
What I Can and Cannot Offer
I will not write downward, simplify until all meaning vanishes, leaving nothing but airy generalizations and a few pictures. But this field is vast, this disease beneficiary of thousands of primary scientific papers. So my best choice is a review of reviews. Accordingly, the linked references are to what seem to me reasonable reviews from recent time. We have contributed two clinical papers I think better than or as good as their like elsewhere, so I have used them for detailed illustration.
Because my goal is kidney stone prevention, always, I write that physicians may diagnose and cure this disease with a greater passion and facility. And if patients who know this curable disease lurks within their ranks seek with greater persistence a proper evaluation for it, that too furthers my desires. Likewise I believe prevention benefits when people understand things. So albeit in a summary fashion I have put in a lot of details.
Outline of Calcium PTH Axis
I drew the sketch pictured below to reveal the mere posts and beams of an elegant mansion.
Serum calcium (SCA) and phosphate (SP) reside in a suitably named rectangle. The red arrows – eg. from calcium to parathyroid hormone (PTH) – connotes down regulation. Blue arrows mean the opposite. For clarity I left out receptors.
For orientation, parathyroid glands number four on average and reside at the four corners of the thyroid gland in the neck. PTH signals kidneys, bone and the gut so as to maintain great stability of blood calcium and a less rigorous stability of blood phosphate concentration.
Low Serum Calcium Increases PTH Secretion
As if to run the system through an accustomed pathway, consider a healthy person who chooses to eat a diet low in calcium.
The GI tract (GUT) has little calcium to absorb, but kidneys lose some calcium in the urine every day. If nothing happened beside low calcium diet, serum calcium would fall. But the parathyroid glands possess a calcium receptor (CaSR) on their cell surfaces – a protein ensemble capable of responding to serum calcium. E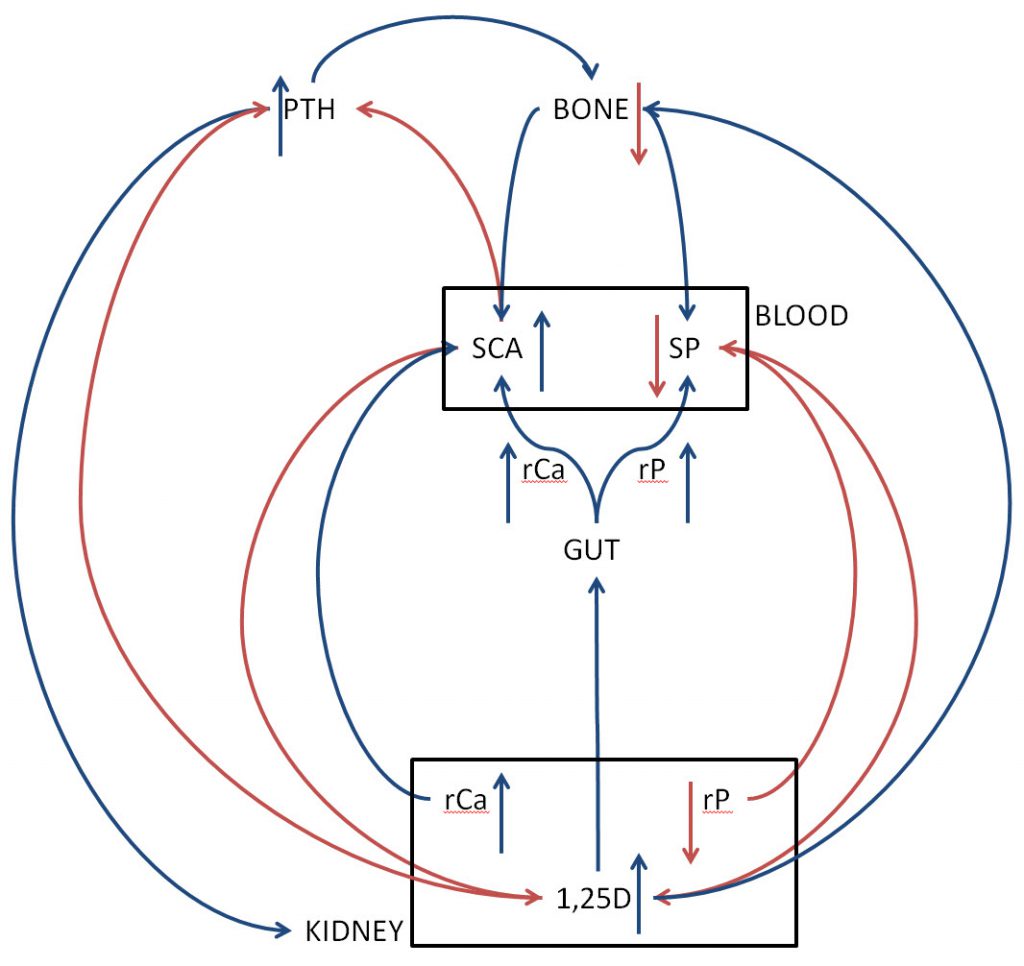 ven a slight fall of serum calcium signals release of preformed PTH in the glands and gradual increase of new PTH production. Here is a fine review of the process.
ven a slight fall of serum calcium signals release of preformed PTH in the glands and gradual increase of new PTH production. Here is a fine review of the process.
Protracted CaSR activation signals parathyroid cell proliferation. With time low calcium diet – as an example – can cause the four parathyroid to enlarge – a form of secondary hyperparathyroidism.
PTH Increases Kidney Calcitriol Production
PTH signals kidneys through PTH receptors to increase production of activated vitamin D, 1,25 dihydroxyvitamin D, or calcitriol. Calcitriol increases the fractions of calcium and phosphate absorbed by the gut (rCa and rP of gut) so more food calcium and phosphate enter the blood. Because of increased PTH signalling I just described, kidneys lose the extra phosphate in urine but retain much of the calcium.
In concert with PTH, calcitriol acts on bone cells to increase mineral losses. This increases bone mineral delivery into blood.
The net effects of PTH on bone and kidney maintain serum calcium normal without an increase of serum phosphate and with minimum increase of serum PTH itself. But bone mineral can be lost.
Calcitriol Down Regulates PTH Secretion
Like other steroid hormones, calcitriol acts via its receptor, the vitamin D receptor (VDR), to alter gene expression. Specifically, calcitriol downregulates the genes for the PTH precursor molecule, and also upregulates genes for the CaSR thereby increasing the amount of the CaSR on the cell surface so the serum calcium signal increases. Consequently, as increased PTH stimulates calcitriol production, calcitriol suppresses PTH forming a negative feedback control loop.
Serum Calcium and Phosphate Down Regulate Calcitriol Production
Just as calcitriol increases them, serum calcium and serum phosphate, independently, both down regulate calcitriol production – another feedback loop. Calcium acts via the CaSR in kidney proximal tubules. Phosphate presumably acts through another hormone, FGF23 (see directly below). Note that on the figure serum phosphate has a downward red arrow that might confuse things by suggesting that a fall in serum phosphate suppresses calcitriol. It is simply that I cannot draw everything in on one figure. PTH reduces phosphate retention by kidneys and lowers serum phosphate. Serum phosphate downregulates calcitriol. The fall in serum phosphate from PTH will reduce that inhibition so calcitriol increases – double negative.
By modulating calcitriol increase from PTH, serum calcium and phosphate stabilize their own blood concentrations.
Calcitriol Down Regulates Itself and Upregulates Kidney Calcium Retention
Calcitriol acts through the VDR in kidney cells that produce it – proximal tubule cells – to reduce its own production. Calcitriol increases production by bone cells of FGF23 – discussed below – that in turn suppresses calcitriol production through its own receptor complex. These feedback loops enables calcitriol to stabilize its blood concentration.
PTH and Calcitriol Increase Kidney Ca and Reduce Kidney P Retention
Calcium Reabsorption
PTH and calcitriol signal kidney to increase the fraction of filtered calcium reabsorbed back into the blood (rCa in the kidney box). At the same time, PTH reduces that fraction for phosphate (rP, kidney). As a result, the kidneys retain the calcium that came into blood from food and from bone but discard the phosphate. This maintains the normal ratio of serum calcium to phosphate.
PTH and calcitriol increase kidney calcium reabsorption mainly at the distal convoluted tubule. PTH down regulates the sodium chloride cotransporter. This reduces NaCl entry into the cells and thereby facilitates calcium uptake. PTH and calcitriol increase the abundance of TRPv5 that permits cell calcium entry. Klotho, a protein made in the distal convoluted tubule removes carbohydrate from TRPv5 delaying removal from the cell membranes.
These hormones increase the abundance of calbindin, a protein in the cell that binds calcium and ferries it to the blood side membrane.
Phosphate Reabsorption
PTH
Through its receptor PTH down regulates phosphate reabsorption by signalling removal of Npt2A and Npt2C from the apical – tubule fluid side – membranes of proximal tubule cells. These two transport protein assemblages account for most of the reabsorption of filtered phosphate. So their removal from the cell membranes permits an abnormal fraction of filtered phosphate to leave the body. The elaborate signalling pathways are detailed in this excellent review. The PTH receptors – proteins PTH attaches to in order to signal cells – are on both the basolateral – blood site – and apical membranes of the cells so the hormone can act from both the blood and tubule fluid.
FGF23
This hormone produced mainly by osteocytes (see below), also reduces kidney phosphate reabsorption but through a different receptor and signalling pathway. It acts like PTH in removing Npt2a and Npt2c from the apical membranes. So both facilitate kidney removal of phosphate liberated from bone or entering from food when PTH increases. Calcitriol and PTH upregulate FGF23 that in turn downregulates calcitriol and PTH production.
If dizzy from all this, think about it as if there were a reason. Higher PTH from, say, low calcium diet will raise calcitriol – more calcium and phosphate absorbed from food and removed from bone. PTH raises FGF23. The two act in concert to lower kidney phosphate retention while FGF23 suppresses calcitriol production – a sort of feedback regulation and both FGF23 and calcitriol downregulate PTH.
The Cell Surface Calcium Receptor (CaSR) Down Regulates Kidney Calcium Retention
I think of kidney CaSR as a parallel regulator of calcium reabsorption acting along with the PTH and calcitriol receptors. PTH and calcitriol act mainly in the distal parts of the nephron. CaSR acts mainly in the thick ascending limb of Henle’s loop, a region that reabsorbs far more calcium.
In humans kidneys cells of the thick ascending limb and distal convoluted tubule express CaSR on their basolateral (blood side) membranes. Activation of thick ascending limb CaSR reduces calcium reabsorption. Distal convoluted tubule CaSR signalling reduced TRPv5 membrane abundance in one study but its overall importance is as yet a matter of debate. Rising serum calcium will through the CaSR reduce kidney calcium retention, in opposition to effects of PTH. As if Nature could not resist yet another layer of control, calcitriol upregulates CaSR in renal tubules as it does in parathyroid cells, further countering the rise in serum calcium produced by PTH.
CaSR in the proximal tubule, on the apical surface (facing tubule fluid) may offset the effects of PTH to reduce phosphate losses. I omitted some of these regulations from my figure drawing to avoid clutter.
PTH Calcitriol and FGF23 Signal Bone Mineral Release
PTH
In concert with calcitriol, PTH signals bone to release mineral, hydroxyapatite. This is the same crystal that makes up calcium phosphate kidney stones and plugs kidney tubules. The extra calcium from bone enters the blood so serum calcium rises back toward normal.
Calcitriol and PTH
Like PTH, calcitriol acts on bone to cause release of calcium and phosphate from bone mineral. The effects of PTH are more rapid, those of calcitriol require hours to days. The two hormones, PTH and calcitriol, each signal production by osteoblasts – bone producing cells – of RANKL which binds to its receptor (RANK) on osteoclasts (bone mineral liberating cells) and osteoclast precursor cells. RANKL recruits more osteoclasts. Osteoblasts also produce a ‘decoy’ protein, osteoprotegerin, that binds RANKL and prevents its action – a kind of internal negative feedback regulation. The net effect is that low calcium diet via increased PTH and calcitriol leads to bone mineral loss via increased RANKL.
But bone mineral loss triggers increased bone mineral formation, so the process mainly acts to speed up bone mineral turnover. However when PTH is high for extended periods of time the net effects are for gradual loss of mineral as the increase of new bone mineral formation lags behind that of bone mineral dissolution.
FGF23
Relation to PTH and Calcitriol
Osteocytes (former osteoblasts caught in the bone mineral they made) and osteoblasts themselves produce this hormone that reduces kidney phosphate retention like PTH does but by a different pathway. Calcitriol upregulates it and, as I already mentioned, it down regulates calcitriol production. PTH also upregulates FGF23 which in turn downregulates PTH secretion.
Regulation By Phosphate
High serum phosphate increases FGF23 production suggesting a phosphate based regulation system not wholly dependent on calcitriol and PTH. This mechanism is clearly evident in people with kidney disease. By contrast patients with PHPT have low serum phosphate.
Regulation of Bone Mineralization
What follows appears to be the contemporary beliefs about specific bone regulatory factors that might be important in PHPT bone disease. I have left out a number of pathways, and warn that much of what I can say comes from animal and cell work and may be untrue in intact human bone in vivo.
FGF23 is centered in an osteocyte network within bone that controls bone formation. Sclerostin, which PTH down regulates, itself down regulates production of the matrix in which bone mineral forms. It also upregulates FGF23 that promotes production of inorganic pyrophosphate a potent inhibitor of bone mineral – hydroxyapatite crystals. The result is low bone mineralization. When sclerostin is low, matrix production rises, FGF23 falls, and therefore inorganic pyrophosphate production falls and bone mineralization increases. Here is an excellent review with pictures. Inorganic pyrophosphate inhibits mineral formation both directly and by stimulating production of osteopontin, another crystal inhibitor, and non specific alkaline phosphatase. The latter normally inhibits inorganic pyrophosphate production – another of the dizzying feedback loops in the mineral system.
Sclerostin, incidentally, may possibly through upregulation of FGF23, reduce calcitriol and thereby urine calcium excretion
A Picture of this Elaborate Web
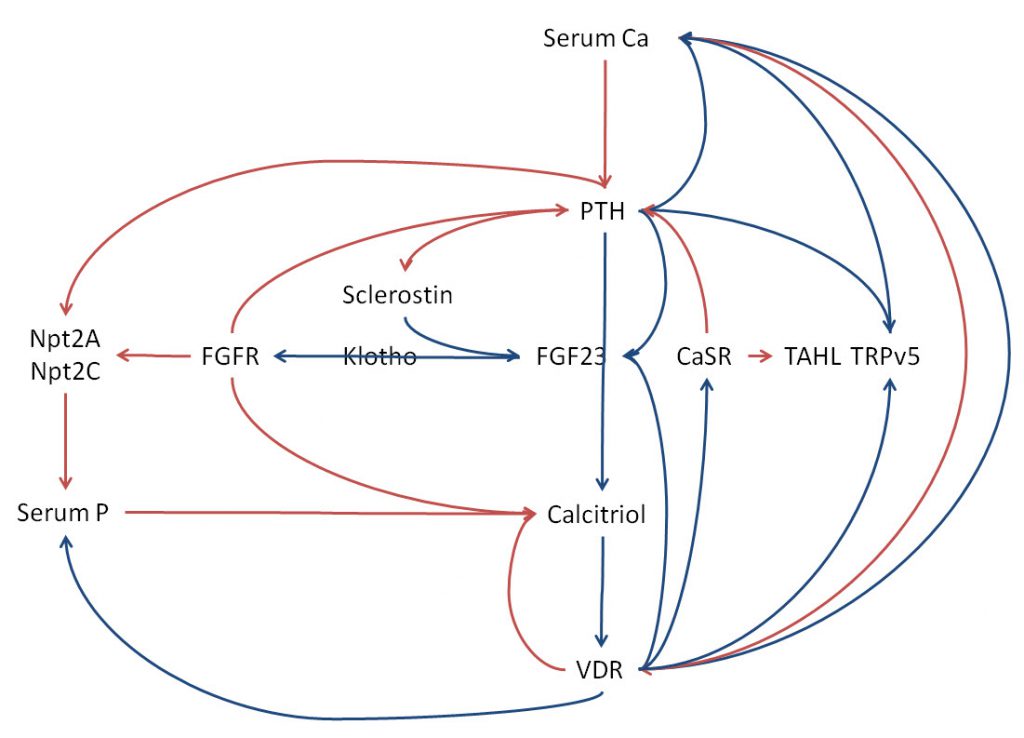 I cannot leave this mass of hormones and their mutual regulations without some try at synthesis. So many names, so many loops. Here, I placed the main names but left our RANKL RANK osteoprotegerin, and osteopontin – no more room. As I have already referenced each regulation step I omit them here. Blue lines show upregulation, red the opposite. Most receptors are left out to avoid clutter – FGFR and VDR are included.
I cannot leave this mass of hormones and their mutual regulations without some try at synthesis. So many names, so many loops. Here, I placed the main names but left our RANKL RANK osteoprotegerin, and osteopontin – no more room. As I have already referenced each regulation step I omit them here. Blue lines show upregulation, red the opposite. Most receptors are left out to avoid clutter – FGFR and VDR are included.
PTH Calcitriol CaSR, VDR
Along the middle is the easy part. Serum calcium down regulates PTH via the parathyroid gland CaSR and calcitriol via the kidney CaSR (kidney CaSR not drawn in along these two lines). Down regulation of calcitriol by calcium is the red curve second from the right. Because I needed the VDR and want to avoid lines crossing , it points to the VDR – incorrect but visually workable.
PTH upregulates calcitriol that through the vitamin D receptor in parathyroid glands up regulates the CaSR that down regulates PTH secretion and down regulates the PTH gene expression – curves from VDR to PTH and FGF23.
Through upregulating kidney CaSR (CaSR is written once, but used for PTH and TAHL) calcitriol downregulates its own production. Through the intestinal VDR calcitriol increases calcium and phosphate uptake from food – long blue curves from VDR back to serum calcium and phosphate. PTH by reducing kidney Npt2A and Npt2C lowers serum phosphate. Through upregulation of the CaSR, calcitriol and serum calcium down regulate calcium reabsorption in the thick ascending limb which reduces calcium retention.
TRPv5
A key entry point for calcium entry into distal convoluted tubule cells, this transporter is upregulated by PTH and calcitriol. That promotes calcium retention.
FGF23, FGFR, Klotho Sclerostin
PTH and calcitriol upregulate FGF23 production which in turn through the FGF receptor (FGFR) down regulates calcitriol and PTH. Klotho, produced in kidney distal convoluted tubule cells is needed for FGF23 to bind to its receptor. Like PTH, FGF23 downregulates phosphate retention by reducing membrane Npt2A and 2C. Because PTH normally downregulates sclerostin which stimulates its production, FGF23 should be low in PHPT.
A Fearsome Corner of Biology
You might have guessed from my drawing and all of the other details I could not draw in that evolution has provided a powerful regulation system for serum calcium so it cannot vary except within a narrow range. That guess seems true. Imagine, then, the havoc and chaos when a key element of the system goes awry. Bone cells, intestinal cells, kidney cells, but most importantly the key signalers PTH, calcitriol, the calcium, receptor. When they become, so to speak, autonomous and regulate exterior to the needs of the entire system, disease results in many parts of the body. In writing this section I came upon this excellent review with clear pictures. Those hungry for a readable scientific paper might like it.
Effects of Low Calcium Diet
Could all this be a dream, some phantasm of troubled minds?
I found this isolated and probably little noticed set of observations and brought them here to illustrate that PTH and calcitriol indeed adapt to low calcium intake and that the remarkable complexity of regulation permits considerable dissociation between the magnitude of their relative change.
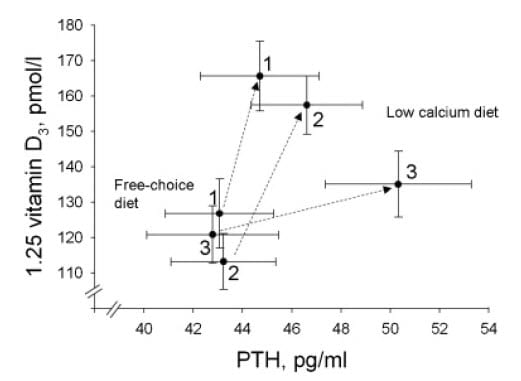 Among calcium stone formers without PHPT or any other systemic disease serum PTH and Calcitriol were measured under proper conditions while on a free choice and after 7 days of a low calcium diet. Figure 3 from the paper shows that among those with well preserved bone mineral density (group 3) PTH rose, as expected and so did calcitriol. Among those with slight or marked reduction of lumbar spine bone mineral density (groups 2 and 1, respectively) the rise of calcitriol was more marked and that of PTH much less so. Serum calcium fell slightly with the diet.
Among calcium stone formers without PHPT or any other systemic disease serum PTH and Calcitriol were measured under proper conditions while on a free choice and after 7 days of a low calcium diet. Figure 3 from the paper shows that among those with well preserved bone mineral density (group 3) PTH rose, as expected and so did calcitriol. Among those with slight or marked reduction of lumbar spine bone mineral density (groups 2 and 1, respectively) the rise of calcitriol was more marked and that of PTH much less so. Serum calcium fell slightly with the diet.
That bone mineral stores somehow condition the calcitriol PTH relationship could seem odd except we have inspected so many links between them and bone. What I have written down does not explain this graph, but the graph is an image, an imago dei. Something found in the real world to symbolize the shocking elaborations of the bone and mineral system.
Clinical Primary Hyperparathyroidism
The modulated and subtle increases of PTH with low calcium diet we call secondary hyperparathyroidism – a response that brings the system back into some semblance of normalcy. Other common reasons for such secondary hyperparathyroidism include insufficient vitamin D and even slight reductions of kidney function.
But what if parathyroid cells choose to grow and proliferate – commit high treason. And, as a result, raise the level of PTH as a primary event? What then?
It is, as I said at the beginning, a remarkable experiment of nature. As if to instruct us nature contrived this singular hormonal disorder so we can see how the system runs. But not for us did nature do this, being indifferent to us as to all that live on Earth. It just happens sometimes. Parathyroid glands lose their normal growth regulation and begin to over produce PTH and grow.
Kidney stone clinicians need special wariness because perhaps five percent of stone patients have PHPT. And, unlike most stone diseases, this one is usually cured by a surgery that modern technology has made surprisingly safe and easy for patients.
Serum Calcium Rises, Phosphate Falls
All factors raise serum calcium. Bone loses calcium and phosphate into the blood. Calcitriol increases because of high PTH. The higher serum calcium suppresses calcitriol, but lowered phosphate does the opposite. So high serum calcium opposes high serum PTH and low serum phosphate. The two outweigh the one. Calcitriol exceeds normal, and increases GI calcium and phosphate absorption into the blood. Kidneys release the phosphate but retain the calcium. Calcitriol would suppress PTH secretion but the glands no longer listen as attentively.
Serum PTH Is Not Suppressed
Although treasonous, the parathyroid glands may long remain not unreasonable. The powerful suppressive effects of increased serum calcium and calcitriol have sway in many people for many years. So serum PTH may stay in the normal range but does so despite elevated serum calcium that would – in a normal person – have driven PTH secretion to its minimum and serum PTH to below the lower limits of normal. For example, a primary increase of calcitriol that raised serum calcium would suppress serum PTH completely. Ultimately, as the gland or glands enlarge more and more, serum PTH rises above normal.
How, you might ask, can the abnormalities of PHPT remain if the very hormone remains normal? Easy. It is normal because of the abnormal serum calcium and calcitriol levels. If they – serum calcium and calcitriol – were normal, PTH would be high.
Urine Calcium Is High
PTH Increases and CaSR Reduces Calcium Reabsorption
I said PTH and calcitriol stimulate kidney cells to reclaim filtered calcium back into the blood, and that is what I meant to say. It does. But that effect is muted.
PTH stimulates calcium reabsorption late in the nephron, at the distal convoluted tubule. But the high calcitriol increases CaSR abundance not only in parathyroid glands but in the kidney and most specifically in the thick ascending limb – a region of major calcium reabsorption. The combination of increased CaSR and high serum calcium reduce calcium reabsorption in the thick ascending limb.
Once again, things balance each other, though not quite. PTH and calcitriol strongly increase calcium reabsorption in the distal convoluted tubule whereas the CaSR reduces it in the thick ascending limb. Overall, if one measures carefully, these opposing factors reduce the fraction of filtered calcium reabsorbed. That alone could increase urine calcium excretion. But another factor adds mightily, and to great effect – increased calcium filtered load from high serum calcium.
Hypercalcemia Increases Filtered Load of Calcium
Kidneys resemble a funnel. At their tops – filtration by glomerulae – they filter about 120 – 140 liters/day of water and salts from blood. Calcium not bound to protein and therefore free to filter measures about 45 mg/liter. So kidneys filter perhaps 5,400 to 6,300 mg of calcium daily. Even with hypercalciuria, urine calcium losses never reach much above 700 mg/day and in stone formers usually average 400 mg/day or less. So marked hypercalciuria reflects perhaps 7 – 9% of filtered load in the urine, normal urine calcium of 100 – 200 mg/day around 1 – 3 percent of filtered load.
Because serum calcium has increased from – let us say – 45 to 50 mg/liter, filtered load increases to 6,000 to 7,000 mg/d. Urine calcium levels run high, perhaps about 350 to 450 mg/day. This gives a range from 5.8 to 7.5% of filtered load in the urine. So the push pull of increased distal convoluted tubule and decreased thick ascending limb calcium reabsorption along with increases filtered load increases urine calcium.
Calcitriol Increases and More Calcium and Phosphate are Absorbed
Bone can lose calcium and phosphate and by doing that support long term increase of urine excretions for both. But in PHPT calcitriol increase from high PTH and perhaps low serum phosphate activates intestinal calcium and phosphate absorption.
Actual Results in Patients
I modestly propose that my own paper with Joan Parks and Elaine Worcester has the prettiest published graphs of data from patients with PHPT. We combined data from 105 patients who had PHPT proven by 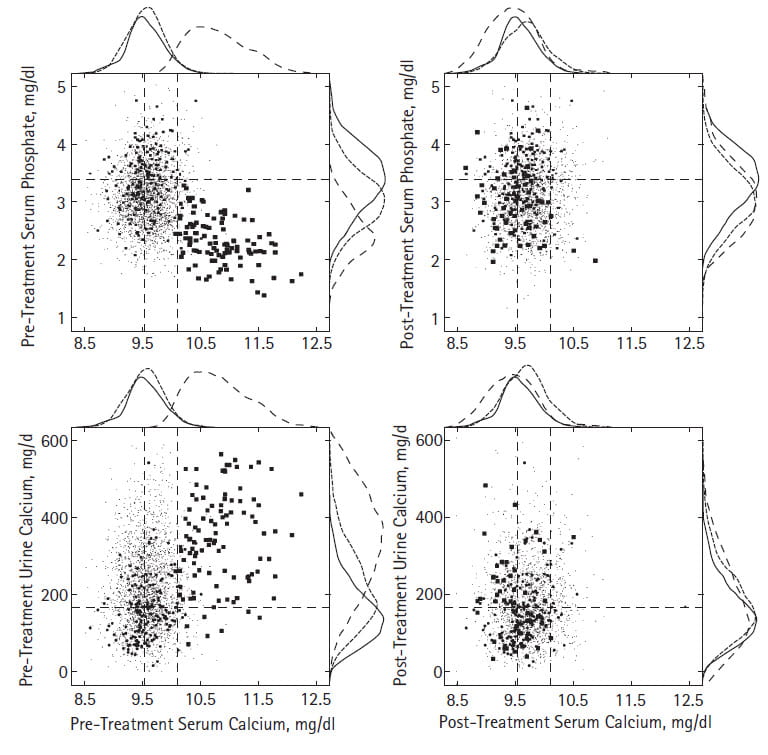 surgical cure with data from 260 normal people and 2416 calcium stone formers.
surgical cure with data from 260 normal people and 2416 calcium stone formers.
Serum Calcium and Phosphate
How The Figure Works
Let me explain this figure to you before you give up and leave the page.
The horizontal axes of all four plots show serum calcium, before (left) and after (right) surgical cure of PHPT. The vertical axes of the top panels show serum phosphate, the bottom panels urine calcium.
Dashed horizontal lines center on the mean normal values for serum phosphate or urine calcium. The dashed vertical lines show the normal mean and upper limit of normal for serum calcium.
Our normals show as mid size dots, common stone formers as a cloud of microdots, and PHPT stone forming patients as large dots.
Because we use atomic absorption measurements not available commercially, our serum calcium values almost never lie above 10.1 mg/dl – the rightmost vertical line.
Here I focus on the two upper panels.
Before Surgery
Serum phosphate falls in concert with rising serum calcium among PHPT patients – upper left panel. Because PTH raises serum calcium and lowers serum phosphate – by reducing kidney phosphate conservation, we expect this pattern. What a treat to observe in real people what theory predicts!
The outer borders support density curves for normals, common stone formers, and PHPT as solid, fine dashed, and coarse dashed lines, respectively. These give a visual impression of how the points distribute. Indeed, common stone formers have lower serum phosphate values than normals, though PHPT far surpasses them. Of course the serum calcium for PHPT is far to the right – higher – compared to the normals and common stone formers.
After Surgery
Only two scattered blood calcium values from PHPT patients lie above 10.1, the upper limit for our laboratory. The PHPT data now overlay data from normals and common stone formers.
Urine and Serum Calcium
Now, take a look at the two lower panels.
Before Surgery
The large dots in the lower left panel show the vigorous hypercalciuria of PHPT and how urine calcium rises with serum calcium. Rising serum calcium means rising filtered load of calcium and rising signal to the thick ascending limb CaSR so theory predicts urine calcium should rise with serum calcium. How nice to observe what theory predicts. No wonder stones occur given how high PHPT raises urine calcium!
After Surgery
After surgical cure, most things subside. But urine calcium remains as high in some cured PHPT patients as in common calcium stone formers. If 200 mg/d is the threshold for kidney stone risk, then about half remain at some risk. This means that cure of PHPT does not complete stone prevention. That residual hypercalciuria needs treatment as if it were idiopathic hypercalciuria. You might say, the border density plots for urine calcium all overlap – normals, PHPT, and common stone formers alike. The three kinds of dots overlap, too. Yes, I would say, but idiopathic hypercalciuria occurs among normal people, and commonly among stone formers. Among stone formers we treat it, among normals we do not. PHPT patients, here, all have been stone formers.
Serum PTH
Because our work spanned 3 decades we worked through the development of PTH assays all with different units and sensitivities. To accumulate what we have measured, we scaled each assay to its 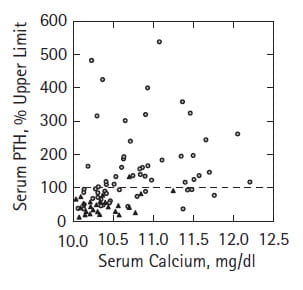 upper limit and plotted the values we had against serum calcium from the same blood samples.
upper limit and plotted the values we had against serum calcium from the same blood samples.
All blood calcium values matched or exceeded 10 – our upper limit in females, but PTH values commonly lay below the upper limit for the assay, shown here as the dashed horizontal line at 100%. These data, that many others have collected in greater abundance and higher assay quality, substantiate the theoretical point that one need not find high PTH values to diagnose and surgically cure PHPT. What counts is higher than normal serum calcium with PTH levels not suppressed.
Supersaturations and Stones
With such high urine calcium excretions how could be expect anything but high urine supersaturations? And we found them.
If anyone has not as yet encountered supersaturation as the main determinant of crystal formation in stone formers – despite our emphasis on it on this site, here is a good review.
Supersaturations
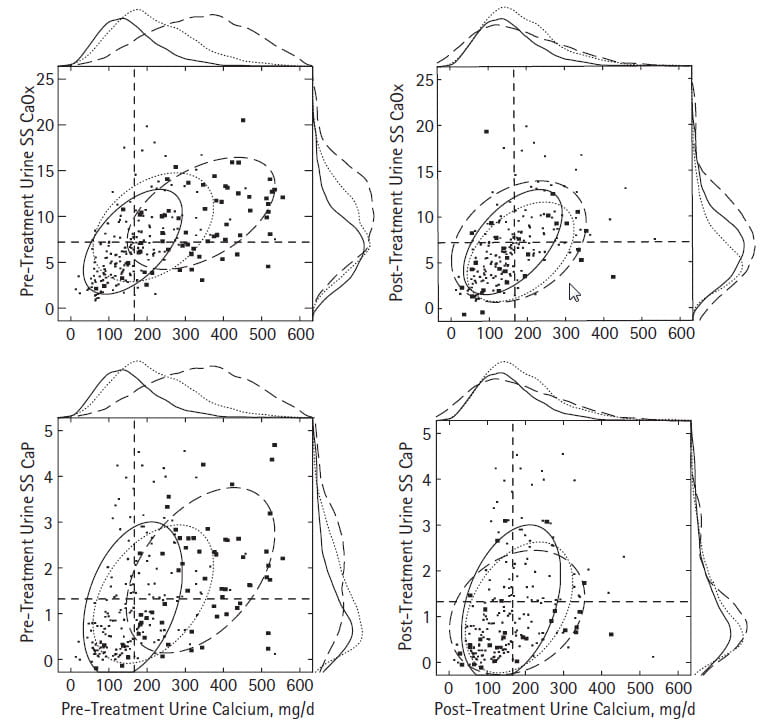 Before surgery, both CaOx and CaP supersaturations (upper and lower left panels) rose with urine calcium excretion. For visual clarity we omitted the cloud of tiny points for common stone formers and show only their confidence ellipse in fine dashed lines. PHPT raises both CaOx and CaP SS above the other two groups.The mean values for both exceed normal and common stone formers – you will find the means and statistics in Table 1 of the article.
Before surgery, both CaOx and CaP supersaturations (upper and lower left panels) rose with urine calcium excretion. For visual clarity we omitted the cloud of tiny points for common stone formers and show only their confidence ellipse in fine dashed lines. PHPT raises both CaOx and CaP SS above the other two groups.The mean values for both exceed normal and common stone formers – you will find the means and statistics in Table 1 of the article.
But on close look that for CaP creeps up higher along the right sided border plot; see where the coarse dashed line bulges in the upper portions.
Stones
From this one might expect both CaOx and CaP stone, and we found them. If we call CaP stones those that contain above 50% apatite, 11% of common stone formers produced them vs. 22% of PHPT patients – a significant difference. But this difference concerned only men; women PHPT and common stone former alike produced about 20% phosphate stones.
The message: CaOx stones or CaP stones make no difference – both kinds of stone former can have PHPT as a cause.
Stone Prevention
Surgical cure abolishes the fascinating mineral abnormalities of PHPT but do stones stop?
We adjusted for the interval between the first stone and surgery and thereafter for the interval from surgery to our last contact with patients, as well as for the number of pretreatment stones. This gave us stones essentially per year of observation. Among PHPT values were 8 before vs. 0.8 after surgery. No one doubts surgical cure reduces stones in PHPT, so our data are just another drop in the ocean.
How Stones Form
Thus far only our research group has presented data about how stones form in people with PHPT. Because each patient study demands great effort and expense, and grant support imposes considerable limits, we could study only five patients. But from them we harvested huge amounts of information. We hope others will do more cases and confirm and extend our work.
You might want to review material about plugs and plaque, and how stones form before plunging into what follows.
Surgical Photographs
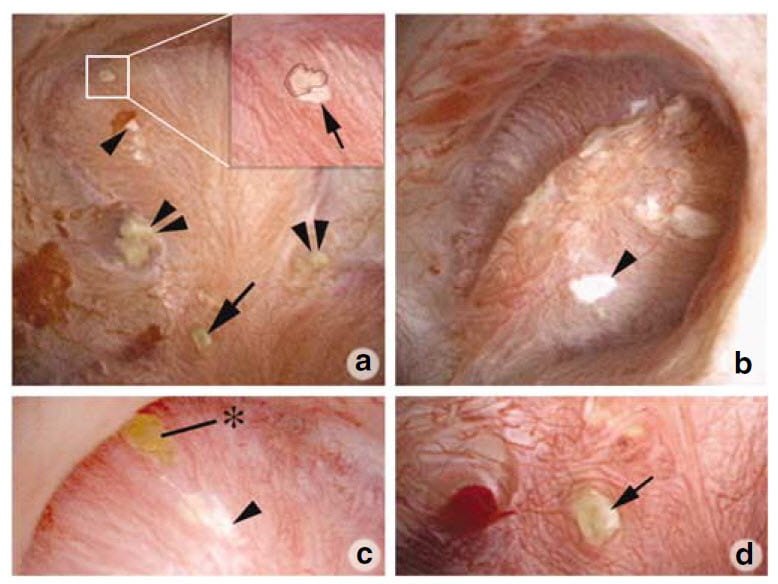 So far as we presently know, stones can form over interstitial plaque or over tubule plugs. We found evidence for both.
So far as we presently know, stones can form over interstitial plaque or over tubule plugs. We found evidence for both.
The upper left panel of this picture shows one papillum from a PHPT patient during stone removal surgery. The white box outlines a small stone on plaque. In the blowup inset to the right, a black line outlines the stone. The single arrow points to the plaque border. A single arrowhead points to more white plaque just below the box. The double arrowheads point to crystal plugs in tubules whose ends protrude into the urinary space. The single arrow at about 6 o’clock points to another protruding plug end.
Panel b shows another papillum from this same patient with a large area of plaque highlighted by the single arrowhead. Yet another papillum (panel c) shows a small attached stone at the asterix just above a large area of plaque. Finally, a fourth papillum (panel d) shows a large plug whose end protrudes quite a way out into the urinary space.
All five patients showed evidence for some plaque and some tubule plugging, but plaque and plug abundances varied widely between patients and even between papillae of a single patient. Accordingly, some papillae seemed almost normal, others very damaged and deformed.
Biopsy Tissue
For 15 years we have obtained tiny biopsies during surgery from those who gave us consent. Often, as here, we studied the tissues by microscopy and also by micro – CT analysis. For this latter, special equipment makes very high resolution CT images of tissue samples – these are very tiny! – that show tubule plugging and plaque.
The left panels below show such a CT image of three plugs within one papillary biopsy sample. A microscopic image (panel b) of the large deposit at the arrow in panel a shows 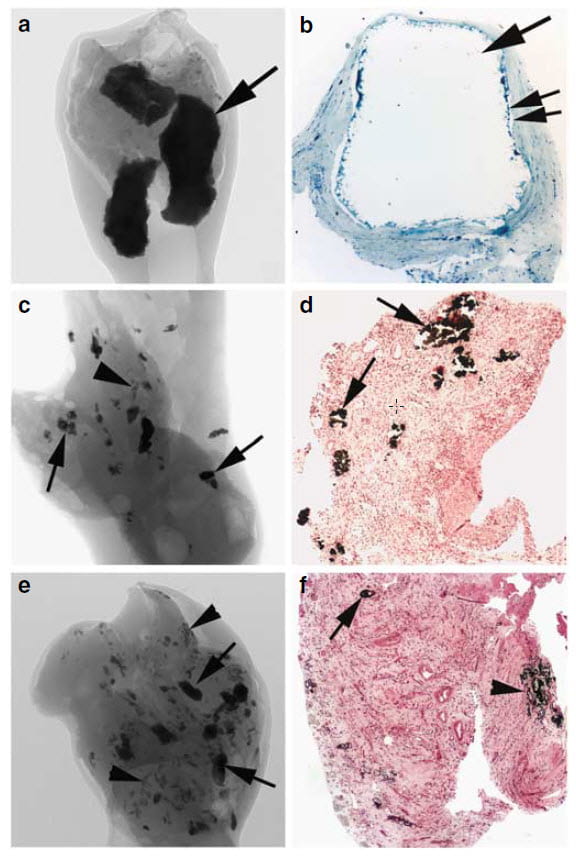 the hugely dilated inner medullary collecting duct that contained the deposit. No cells line the duct; they are all dead. Presumably plugging killed them.
the hugely dilated inner medullary collecting duct that contained the deposit. No cells line the duct; they are all dead. Presumably plugging killed them.
Panel c shows both multiple smaller plugs by CT that arrows point to and some plaque at the single arrowhead. In the histology section – panel d – the arrows point to plugs. Similarly, but with more abnormalities, panel e shows many deposits (arrows) and regions of plaque (arrowheads). The histology (panel f) shows a plug at the arrow and plaque at the arrowhead. The tissue is partly scarred.
In all cases, hydroxyapatite crystals comprise both the plaque and plugs.
Damage Can Be Extensive
I should not burden this article with yet more pictures but simply say that compared to the idiopathic calcium stone formers patients with PHPT most resemble those with calcium phosphate stone. They, too, have many plugs, loss of lining cells, and papillary deformities.
But PHPT goes further and damages more. Deposits reach up from the medulla all the way into the cortical collecting ducts. The cortex of kidneys contains the crucial glomerulae and complex proximal and distal convoluted tubules. Damage in that region tends to cause a loss of kidney function.
PHPT Can Reduce Kidney Function
In principle, any stone disease can damage kidneys by obstruction or requiring surgical procedures. In fact, large population surveys show reductions of kidney function as a general trait of stone formers in general. But PHPT reduces kidney function more, and with greater regularity than we find in the common idiopathic calcium stone patients.
PHPT also associates with vascular disease and reduced longevity. This is no place to detail such broad issues that many reviews cover very well indeed.
PHPT Exposes Kidney Cortex to High Calcium levels
Keep in mind that PHPT imposes on kidneys both high blood and high urine calcium. The latter occurs in routine stone formers, the former does not. The medullary collecting ducts have a sturdy character. Because of water conservation, tubule calcium concentrations can be very high in even normal people. But glomerulae, proximal tubules, and even the distal convoluted tubules do not normally encounter high calcium levels. They have a delicacy about them. Injury to these cortical structures can bring on kidney disease and even kidney failure.
Diagnosis Of PHPT in Stone Formers
Whereas borderline or frankly elevated serum calcium levels pose diagnostic problems in general, among calcium stone formers the range of causes for such levels narrows. In my fifty years of kidney stone prevention work diagnosis of PHPT and separation from look alike diseases has cost me little trouble.
Hypercalcemia
The cornerstone of diagnosis deserves meticulous attention to all details.
Blood Management
We draw all our bloods fasting, in the morning. In our CRC studies we documented modest changes in serum calcium with food that could complicate diagnosis.
PTH levels in plasma remain stable whereas those in serum do not. Therefore we prefer plasma samples for PTH assays. We often say ‘serum PTH’ but I usually obtain plasma PTH levels.
We are among the few who use atomic absorption calcium measurements standardized using aqueous standards. The method narrows the variability of the assay and permits diagnosis in patients with very modest hypercalcemia. But modern high throughput instruments can suffice if laboratories take care to standardize carefully and establish a normal range.
Having this technique has spoiled us. We shun common aids to diagnosis such as ionized calcium and albumin corrected serum calcium. They may help others.
Evaluation of Results
In a calcium stone former, even one serum calcium above the normal range for the laboratory in use prompts me to order more. The cost in time and test charges dwindles to nothing at all compared to a missed diagnosis and opportunity for surgical cure. Put against the tens of thousand of dollars paid for even a single kidney stone surgery, or the many thousands of dollars for a mere ER visit, how can one stint on blood measurements that in total could consume perhaps a thousand to two thousand dollars at the most?
With multiple samples, in most patients, one finds stable average calcium values above normal or not. I am personally unrelenting and pursue serum calcium evaluation as long as I need to until I am satisfied.
I do this because serum calcium varies from day to day in patients with PHPT and one may need 10 or even 15 measurements or more in some cases to be sure.
Drugs
Very many drugs can alter serum calcium. Famous ones include thiazide diuretics, lithium, and vitamin D excess that raise it, and loop diuretics like furosemide that lower it. Textbook chapters and reviews list all possible candidates and physicians refer to them. The crucial idea for a broad audience is this: Suspect any drug. If possible evaluate for PHPT in the absence of drugs. Never eat before blood sampling; that includes coffee and especially coffee creamers. Stop any calcium supplements, herbs, and vitamins at least a week before evaluation.
PTH Not Suppressed
Normal or High Values in PHPT
This must be measured in the same blood sample as calcium. In PHPT patients PTH levels can be normal or high. Unlike hypercalcemia whose ascertainment may require many blood samples, just a few PTH values in the normal range or higher suffice.
Normal or High Values in Other Conditions
A few conditions raise serum calcium, and do not suppress PTH. Familial hypocalciuric hypercalcemia (FHH), a genetic disorder of CaSR functioning, does this and separates from PHPT in having a low urine calcium excretion. Lithium treatment and hyperthyroidism can mimic PHPT.
FHH offers an insight into the importance or lack thereof of the CaSR apart from kidney and parathyroid gland. Heterozygotic patients with inactivating gene mutations have hypercalcemia, normal or high PTH and low urine calcium excretion, as I described, and yet display no other significant abnormalities of muscle, balance, or quality of life. Perhaps one good gene copy suffices in other tissues – such as intestine. Certainly, knockout of both alleles in mice causes a severe disorder much like PHPT. Correspondingly, in humans homozygous inactivating mutations of the CaSR cause neonatal severe PHPT.
Association and clinical trial data do not support an important role of CaSR abnormalities in vascular disease. Proposed roles for CaSR genetic variants as causes of kidney stones have not been convincing.
PTH Suppressed
If suppressed, something other than PHPT has caused the problem. Malignancy, granulomatous diseases, excessive vitamin D ingestion, and CYP24A1 deficiency come to mind.
Urine Calcium Not Low
One expects elevated urine calcium excretion as a common trait given stones and PHPT although in fact patients with just bone disease and no stones had urine calcium levels no different from those with stones. Our own data show very few patients with normal urine calcium levels, and these were older women with modest renal function impairment. Measurement of urine calcium will disclose the uncommon FHH patient as FE calcium will be very low – usually below 1%.
Undetermined Patients
In our own report we present patients for whom we could neither recommend surgery nor completely rid ourselves of suspicion that PHPT lurked just beyond our reach. Many went on with little difficulty for years, treated like common stone formers. Only a certain wariness separated them, in my mind, from all the rest.
Normocalcemic Primary Hyperparathyroidism
I know many papers describe normal serum calcium with high PTH as a form of PHPT but personally doubt the usefulness of such a diagnosis in calcium stone formers. Unlike the numerous asymptomatic PHPT patients one encounters in general practice, our patients form calcium stones. Unwilling to operate without hypercalcemia, I treat their stones as I would any idiopathic calcium stone former. In all my years of work almost none went on to frank hypercalcemia. But a few did. So I believe that among the many people who present this way hide a few in whom PHPT smolders.
As a corollary, some patients with PHPT can easily pose as idiopathic calcium stone formers. Even marked hypercalciuria can be idiopathic in such people. Therefore, we need to measure fasting serum calcium in stone formers more or less yearly. Whenever serum calcium seems to rise, that can be a clue that underlying PHPT has become manifest and deserves serious attention. Cure far surpasses even the best management.
Low Calcium Diet
Among 118 people who in a Norwegian population survey had a PTH level above normal. Of these, 82 remained as candidate for additional studies 3 years later. Of those with persistent high PTH, one man and 7 women had developed hypercalcemic and diagnosed with PHPT. In 56 others PTH had remained high, and in 26 it had fallen to normal. Both groups – stayed high, fell to normal – were compared to 131 well matched people from the original cohort whose PTH levels were normal.
Compared to the normals, the people with persistently high PTH had slightly lower serum calcium levels, and by food table analysis lower calcium intakes (400 vs. 592 mg/day). Moreover, their blood pressures were higher (154 vs. 143 mmHg systolic). Only the women, not the men, displayed the higher blood pressure. Although bone mineral densities did not differ in general, among those with persistent high PTH spine bone mineral density was lower when adjusted for quartiles of BMI.
A Coherent Clinical Entity
Reputable mineral research groups have reported cases, mainly older people with bone mineral reduction, whose PTH levels are high and whose serum calcium levels are always normal. In this review of the Columbia University experience of 37 patients who met these criteria, 95% were women, 84% of the women were menopausal, and 73% had low bone mass. None had reduced serum 25 vitamin D nor reduced kidney function.
Over 4 years 22% developed frank PHPT. Their upper limit for serum calcium of 10.4 mg/dl, however, seems high. Although ours is very low because of atomic absorption as our technique, many commercial laboratories consider values over 10.2 mg/dl as an upper limit. Of interest in this point, those who developed PHPT tended to have the highest serum calcium levels.
A review of this subject, that includes references to the 2014 International Workshop on asymptomatic PHPT dutifully reviews the many exclusions needed to fully define high PTH and normal serum calcium as a reliable clinical entity: low vitamin D, any intestinal cause of calcium malabsorption, reduced kidney function, absence of idiopathic hypercalciuria, and exclusion of drugs at the times of measurements. Neither review mentions the obvious – low diet calcium intake that can produce exactly the picture described in these patients. Kidney stones are frequently present and bone mineral loss and fractures. Some patients have come to parathyroid surgery.
My View in Stone Disease
To me, the entity could arise from low diet calcium. I would not at this time recommend surgery but rather high diet calcium, with suitable low sodium intake or even thiazide to moderate urine calcium losses. The patients are mostly older women and some appear to have PHPT with borderline serum calcium levels. Given the generous upper limits mentioned above, I am not surprised that some – those with the higher values – simply had PHPT. I treat and wait; if serum calcium rises, I recommend PT surgery.
Bone Disease
Bones Lose Mineral
Serial measurements can show a gradual fall in bone mineral density of the spine and forearm over time. Of these the distal radius is most affected, spine the least so. The loss of bone is more proportional to serum calcium than to level of PTH. After surgical cure patients restore their bone mineral. Although not essential in stone formers, I and most others evaluate bone mineral in PHPT patients.
Sclerostin
What goes wrong with bone – beside the obvious loss of bone mineral occupies considerable modern research talent. One abnormality concerns sclerostin, a hormone I have mentioned in regard to the multi-hormone loops of mineral metabolism. Osteocytes – bone cells – make sclerostin that inhibits bone formation by osteoblasts – the bone cells that produce bone. PTH is thought to inhibit expression of the Sclerostin gene. Not surprisingly serum sclerostin levels in PHPT lie below those of normal people. This alone would lead to increased bone formation, not loss of bone mineral, so more must be disordered in PHPT.
FGF23
As noted above in earlier sections this hormone is increased by both PTH and calcitriol and increases local inorganic pyrophosphate synthesis. Pyrophosphate inhibits crystallization and new bone formation.
Effects of CaSR
The dynamics of bone mineral regulation gleaned mainly from animals posit a role for the CaSR. Mature osteoblasts produce bone matrix that mineralizes much like kidney stone do but in a productive and controlled manner. When prompted to produce bone these cells produce RANKL (R) so that the ratio of R to its ‘decoy’ ligand (that captures R and prevents its biological actions) osteoprotegerin (O) rises. Free R stimulates formation of osteoclasts that break bone mineral down. CaSR stimulation in osteoblasts can reduce or increase the R:O ratio diminishing osteoclasts and fostering bone formation, or, in older animals raise that ratio so that bone mineral loss rises with formation – coupling. In trabecular bone – like vertebral bodies – mineral can increase while in cortical bone – wrists, for example, it can fall.
Systemic Abnormalities
Common symptoms include constipation, and various mental and emotional changes that can resemble depression. Surgical cure may reduce the neuropsychiatric manifestations of PHPT.
When serum calcium is very high, one encounters loss of appetite, even nausea and vomiting, polyuria, dehydration and a vicious cycle leading to increasing serum calcium and kidney failure. But this course of events is very rare in routine PHPT. I have never encountered it in my stone practice.
Reduced kidney function has been repeatedly documented in PHPT. In asymptomatic PHPT falling function has been considered a reason for surgery. Among stone formers with PHPT surgical cure is a primary aim.
What Causes PHPT
Essentially enlarged glands in all forms of PHPT reflect neoplasia – cell proliferation that creates tumors that are for the most part benign but can occasionally possess the traits of malignancy.
As glands enlarge they exhibit reduced sensitivity of to serum calcium due, probably, to a reduction in the abundance of the CaSR. This leads to increased PTH secretion. Because basal PTH secretion by parathyroid cells remains above zero whatever the level of serum calcium, simple increase of cell number can progressively raise PTH secretion and serum PTH levels.
In 85% of PHPT cases parathyroid glands form a single adenoma – benign enlargement. In 15% of cases multiple glands enlarge – so called hyperplasia. Carcinoma is rare, at less than 1% of cases.
Familial PHPT
A large review of familial PHPT syndromes has been compiled for the European community and those interested might wish to read it.
Reviews quote about a 10% figure for PHPT cases that have a familial – genetic basis. A recent review lists FHH, under the more exact name FBHH, as a form of this disease as it is familial and consists of increased serum calcium with normal or increased PTH. We have already discussed it in relation to its cause in defects of the CaSR and I will not repeat the matter here.
Multiple Endocrine Neoplasia Type 1 (MEN 1).
This well known genetic disease accounts for about 20% of familial PHPT. The affected people inherit one defective gene copy but if the parathyroid cells lose the other as a somatic mutation they develop parathyroid tumors that overproduce PTH. Carriers with one normal copy seem normal. The MEN1 gene codes for menin, a nuclear protein that suppresses cell proliferation. MEN 1 gene deletions cause not only four gland enlargement PHPT but also a mixture of islet cell and pituitary endocrine tumors and endocrine tumors of the duodenum that produce the hormone gastrin that stimulates gastric acid production and fosters peptic ulcer. Other tumors include adrenal glands, thyroid glands and benign lipomas – fatty deposits beneath the skin. PHPT is usually the first manifestation of MEN 1 gene abnormalities.
Multiple Endocrine Neoplasia Type 2A (MEN 2A).
This complex disease arises from mutations in the RET proto-oncogene. Patients have mixtures of thyroid cancer, catecholamine producing tumors of the adrenal glands – pheochromocytoma, and PHPT. But PHPT is present in only about 35% of cases and the diagnosis is made because of the other two tumors. It does not therefore much concern us here.
Multiple Endocrine Neoplasia Type 4 (MEN 4).
This consists in parathyroid adenomas or four gland hyperplasia associated with pituitary, thymus, and adrenal tumors, and tumors involving the stomach and endocrine pancreas. It resembles MEN 2A. It arises from defects in the CDKN1B gene.
HPT-JT
This autosomal dominant condition includes parathyroid adenomas – single or multiple enlarged glands but not all four glands – and parathyroid cancers, along with fibrosis tumors of the jaws, kidney tumors and cysts, and uterine tumors. It arises from mutations in the HRPT2 gene that produces a protein called parafibromin that inhibits cell proliferation. Like MEN 2A this disease is so colorful one diagnoses it readily and cannot confuse it with commonplace PHPT of kidney stone formers.
Familial Isolated PHPT (FIHP)
This consists in what the name says – families afflicted with PHPT that do not harbor any of the other gene abnormalities in obvious forms. It accounts for about 1% of PHPT cases and appears at an earlier age than most PHPT. Diagnosis depends on exclusion of gene abnormalities and clinical findings of MEN 1 and HPT – JT. Among cases in series, some patients have had abnormalities of MEN 1, CaSR and HRPT2 genes. The latter have especially correlated with parathyroid cancer or cystic parathyroid tumors.
Sporadic PHPT
Non familial PHPT arises from isolated single adenomas in 85% of patients and multiple gland enlargement in the rest. They are benign neoplasms that can grow progressively larger but almost invariably remain benign in lacking a potential for spread beyond the gland capsule.
Adenomas
Unlike the familial diseases gene abnormalities are not inherited but arise as somatic mutations. They mainly affect CaSR functioning, but also genes associated with familial syndromes such as MEN 1 and CCND1. Although adenomas may exhibit reduced CaSR abundance and therefore signalling sensitivity, no gene abnormalities of the CaSR or its signalling pathways have been found.
An alternative pathway to adenoma may be epigenetic abnormalities involving aberrant hypermethylation of tumor suppressor genes. Some recent studies have disclosed microRNAs. This excellent review provides references for all of what I say here.
Hyperplasia
This term has a blurred usage in practice. Few criteria reliably distinguish between adenoma and hyperplasia in a single gland. So the term hyperplasia usually stands for multiple enlarged glands. When multiple, adenomas may have a greater propensity to recur – normal glands enlarge later on and require another surgery. Often, if careful, familial PHPT will become apparent from family history. But at least half of multigland enlargement is non familial.
Surgical Treatment
I omit here any review of preoperative imaging or of surgical approaches or surgical pathology as too far outside the goals of this site. Suffice that skilled endocrine surgeons are best for this kind of work as opposed to more general surgeons. My own career experience has been with endocrine surgeons specially skilled in parathyroidectomy. Given that the surgery has considerable complexity and cure is a common expectation, and also given that parathyroid surgery for PHPT is not a particularly common clinical activity I strongly support the idea of using surgeons whose career has centered on such work.
This recent review discusses imaging and surgery. It summarizes the Fourth International Workshop on PHPT that focused on guidelines for asymptomatic PHPT but includes comments about surgery and imaging as a guide to surgery.
Treatment Outcomes
I mentioned our own published data in an earlier section. A review of surgical outcomes of PHPT stone formers reports only one patient with a new stone over 100 months of followup. Another reports virtually no real new stones but only passage of prior stones from kidneys. A very recent review of PHPT summarized multiple observational studies all much the same. Although we have no prospective trials of surgery vs. medical management of stones in PHPT the observational data and the routine nature of curative surgery makes such a trial unlikely and perhaps no longer doable. The prime meaning is that surgical cure is ideal for patients who form calcium renal stones.
On the other hand, another, meta analysis, review found that recurrence rates of new stones after cure remained elevated compared to background rates of new stones in normal control populations. Even so the authors agree that surgical cure benefits stone disease but that postoperative management against new stones should continue. We have found much the same, in that residual hypercalciuria needs management. I illustrated this residual problem earlier.
What Does It All Come Down To?
Here you are, at my final say on PHPT.
Look for it in every calcium stone former, and look with diligence and a suspicious eye. For PHPT is a systemic disease with real potential to harm people that can be cured in most by modern and highly effective and safe surgery. Whereas many older women who have PHPT without any apparent consequence may never need a surgery and do well, no one recommends against surgical cure for stone formers.
Even if normocalcemic, as most are, calcium stone formers may harbor among their ranks a few smoldering normocalcemic PHPT patients who will manifests their disease only over time. This means I favor yearly fasting blood tests that no one thus transformed misses their chance for a cure.
Exclusion of all systemic diseases begins all stone prevention efforts. Frankly it is a fool’s errand to treat any calcium stone former without first excluding systemic disease as the cause. This requires serum and 24 hour urine testing and significant clinical ability. True, a majority will have nothing but idiopathic calcium stones, and whatever money and time used was used to no direct gain. But even one PHPT patient left untreated, even one, could in a year or two of unbridled stones mount up surgical costs in the tens of thousands of dollars. And miseries and dangers too. Even more, that patient’s disease can probably be cured.
Worse happens if one through lack of proper testing misses even more damaging diseases like severe hyperoxaluria. Acute kidney injury and even irreversible kidney failure wait on such patients from even incidental dehydration.
For these and other reasons I am opposed utterly to those clinically naive statisticians who advise all calcium stone formers be treated without blood and 24 hour urine testing. No idea is more likely to cause some patients real harm and, although I hesitate to mention the vulgar, some unlucky physicians their unwelcome day in court.
Thank you SO much for this one!!! I started passing stones in August of 2014, and I’ve seen doctor after doctor. Finally 2 years later and after several surgeries to remove stones, I’m told I have Primary Hyperparathyroidism. Then I’m told its not that, because my calcium is almost always normal, so possibly Normacalicum Hyperparathyroidism. So then I’m sent to another Specialist, more test. And ONE time my PTH test is normal in two and a half years so I’m sent BACK to my kidney specialist and he didn’t agree with two of my other doctors. He says I DO have Primary Hyperparathyroidism BUT…NOTHING about having the surgery I need to fix this. I’ve found not many doctors know about this and we, the patients, are left to suffer!!
Thank you again!!!
Hi Christina, The article more or less summarizes how things work. If your blood calcium is above normal and PTH is not below normal and your urine calcium is not low but high you have primary hyperparathyroidism. If you have high PTH levels but normal serum calcium and I presume high urine calcium the best approach is to be treated as idiopathic hypercalciuria. Normocalcemic primary hyperparathyroidism can be treated as idiopathic hypercalciuria to prevent stones; if it turns into high calcium hyperparathyroidism plenty of time to consider surgery. This is a complicated disease. You cannot help yourself by yourself, just consider these general maxims and let your physicians decide that best to do. Regards, Fred Coe
Dr Coe,
Another wonderful piece that makes the complicated understandable, without simplifying. I am interested in your opinion in the clinical success of parathyroidectomy for nephrocalcinosis stone formers versus renal collecting system stone formers. In Albright’s original papers he notices the reduction in stone formation to population levels but notes that the procedure appears to truly make a difference for the nephrocalcinosis patient. Do you think there are different phenotypes for the disease? I’m never sure I’m doing the right thing if I find elevated PTH in a 50 something first time stone former. The downside from low PTH is not insignificant. Thank you for your continuing posts.
Hi Alex, I have not published on the varieties of stone disease, so I guess I do not know the answer. I did not see any papers along the way in writing the article. But things may be different in the modern age because in his time collecting system stones had poor surgical approaches whereas we have great ones so clearing stone burden after cure of PHPT could make an end of stone events. No trials so far but one could be valuable. As for PTH values above normal without hypercalcemia, I wrote what I believe the papers tell us. Some of the people are early stage PHPT and serum calcium will rise – time enough to operate then. While waiting, I lower urine calcium medically and find stone prevention seems reasonable enough. Operating on normocalcemic patients is not acceptable to me as I am afraid so many factors can raise PTH the surgery would be meddlesome and even harmful. Glad you find the site worthwhile, and thanks for the great question. Warm regards, Fred
In May of 2017 I had shock wave lithotripsy to blast a stone from my kidney. They were calcium stones( 3 types with the majority oxalate). In routine blood test from 2014 to the present blood calcium ranged from 10.2-11.5 with the most recent at 10.2.A June PTH test was 71.1. My urologist ordered the 24 hr urine test and calcium levels were 707. My urologist referred me to a nephrologist suggesting that I might have a parathyroid problem. In the first consultation(Thursday August 17), he recommended that I stop taking vitamin d, as he was sure this was the cause of all of my high calcium levels. My Vitamin D levels vary from the high 50’s to the mid 60’s. I have had several urine infections since March and was given ampicillin. On Friday August 18 I developed another infection that had a burning urination, foul odor and a 103 degree fever, I am on keflex for this infection and have passed several stones since. From 2002-2004 I had the same issues , along with an enlarged prostate. After a TURP all stone formation and infections stopped. At that time I was given keflex, cipro, and levaquin for the infections. I am not looking to repeat this drug progression for my current condition. My Nephrologist ordered blood and urine tests but not PTH. I thought that was unusual as the test is simple and covered by insurance. He believed that this test was not necessary at this time. I was of the belief that the more relevant data a doctor collects the better the chance of a proper diagnosis of the stone problem. Are we taking the proper steps to address this problem, or are there other things to look for. I am not looking to have continuous infections and kidney stones as this has reduced the quality of life.
Hi Douglas, Although I am far away and not fully aware of your entire medical condition, I can make some comments your physicians might find useful. The combination of high serum calcium – 10.2 – 11.5 mg/dl with PTH of 71 and urine calcium of 707 mg/day more than strongly indicates primary hyperparathyroidism. Pathological vitamin D excess can indeed increase serum calcium and urine calcium but will suppress serum PTH via vitamin D effects on Pre ProPTH gene expression and increase in PT calcium receptor abundance. To achieve this one needs a serum 25D level considerably higher than 60 and given the ample serum PTH vitamin D intoxication seems unlikely. I would think one might want to proceed with further evaluation for PHPT with hope of surgical cure, especially given the very high urine calcium and modern non invasive surgical approaches. Probably your nephrologist has already thought along these lines and omitted the PTH level because he already knows it is not suppressed so all emphasis is on serum calcium. Regards, Fred Coe
Dear dr. Coe,
I’ve suffered from calcium stones for a while now and my dr. doesn’t understand why. My PTH is always too high, my blood calcium level is always normal to low-normal and my calcium in urine is almost always high (but one time it was normal). My vitamine D level is normal, so that can’t be the cause. And the dr. tells me that it can’t be the kidney itself that causes this, because the calcium level in urine should be too high all the time and that is not the case. He tried thiazides, but I got sleeping problems and a high heartrate during the night, so he thinks it’s not wise to start the meds again. He now wants to speak with other doctors to see if they think I might have a form of hyperparathyroïdism and if I should have an echo of these glands. I am a lactose intolerant so my calcium intake was low for years. The last months I tried really heard to reach an intake of 1000mg calcium a day, but that had no effect on my lab results (yet). I’m also diagnosed with IBS and hypothyroïdism. Meanwhile I’m so tired and I feel something is just not right. Could you please give me some advice on what to do?
Kind regards,
Liz.
Hi Liz, low calcium diet increases serum PTH. This is part of the adaptation to low calcium diet. Since you appear to have idiopathic hypercalciuria – normal serum calcium, high urine calcium – the increase of PTH can be actually less marked than in normal people, but it can occur. This is called secondary hyperparathyroidism. I have to take on faith that your serum calcium is always normal, of course, and your physician would see to that. Usually serum 1,25D – not the common 25D – is high as well in your kind of situation. Another common cause is very mild reduction of kidney function – eGFR in the range of 60 – 90. Lowering PTH with high calcium intake can take years. If eGFR is even slightly reduced, the level may never come down fully to normal. Surgery is out of the question. Regards, Fred Coe
Good Morning Sir,
Just a follow up on last tests by kidney doctor. Thank you for all that you do to help us understand what is happening to us. PTH is 32, and calcium 9.5. Calcium Urine is 250. have 40 stones in the lining of my kidneys according to CT scan, one year ago two stones, removed Nov. 2016. Serum Calcium 10.0. In 2011 PTH was 76 Calcium 11, had one hot parathyroid removed. Vitamin D is staying in 20’s. Have Fibromyalgia, All but 4 teeth left,osteoporosis , osteoarthritis and kidney stones since 2006 with three operation removal. I am a 60 year old mother and grandmother, what is the solution to the problem here? Oh Urine Uric Acid 0.801. I have appointment Dec. 29th with thyroid doctor in N.C. what will I have to bring to the doctors? I have three KUB’s and two CT scan’s in two years.
Sincerely,
Annette McNamara
Hi Annette, given your normal serum calcium you do not have primary hyperparathyroidism. But your urine calcium is high and I suspect that is helping to form your stones. I suspect a low calcium diet, and that might worsen your bone disease. Ask your physicians if you indeed have idiopathic hypercalciuria; many patients treated for PHPT also have it, and remain hypercalciuria as you are. Regards, Fred Coe
Hello again Dr. Coe, MD
I found this post and thought I would check in with you. Still not much change. Praying Duke will accept me. As my dipstick urine has gone from 6 to 8.5. Parathyroid number 76, and 9.0 on serum cal. Vitamin D 15. Never giving up. Just don’t get doctors that chase the Vitamin D, telling me to take 5,000 vitamin D a day, Thanks for listening. Just praying for a miracle I guess. From 2006 till now all the doctors worry about is the D. http://www.parathyroid.com/hypercalcemia.htm
Hi Annette, The site is commercial. Much of it is medically trivial and can be found in the most elementary textbooks in a better form. Some things are lacking nuance. For example, it is not true that high serum calcium and normal serum PTH means primary hyperparathyroidism if, for example, 24 hour urine calcium excretion is extremely low. That latter means familial benign hypercalcemia and surgery is improper. Put another way, no physician should ever practice from this kind of oversimplified and poorly referenced source. As for you, your serum calcium is normal and the high PTH is not from primary hyperparathyroidism but some other cause. For example your serum vitamin D level is 15 – a low value – and that alone can raise serum PTH. The urine pH over 7.9 suggests infection with organisms that can raise pH by producing ammonia from hydrolysis of urea – your physician can help with this. Duke is outstanding, and they will no doubt consider the high PTH as some reflection of low vitamin D, or perhaps subtle kidney impairment, or perhaps low calcium diet – as I do. Regards, Fred Coe
Very illustrative and I thank you for taking such time. My situation has been complicated and is worsening with little expertise to assist in “management”. I had a missed diagnosis of PHPT due in part to my age early 30’s at the time. I had significant symptoms throughout a triplet pregnancy that was not without its severe complications due almost fully to continued missed diagnosis. 3 years later an indisputable sestamibi revealed the ugly gland and I was finally scheduled for immediate surgery despite >5 years of calcium >10.8 PTH 65 ( intraoperative showed 130) and urine CA 380-500mg. I am now left with continued hypercalciuria of 350-480, 25 oh d 23-27, 1,25 d 60’s, PTH 20-40, total serum CA 7.4-9.2 ionized CA always low and lower than expected in comparison to total. I have low blood pressure, daily bouts of hypocalcemic symptoms with severe bouts with severe arrhythmias (ER initially confused for MI event until metabolic labs came back) All I am left with is micromanaging my d supplementation, calcium, magnesium etc. I take the minimum I can to keep the low CA symptoms away without renal symptoms returning in small spread out doses. All I hear is d is too low with no regard to renal health let alone my continually deteriorating constitutional health. Any suggestions or thoughts on how PHPT though surgically resolved could result in such profound and unexpected results? I am currently 43, no alcohol, healthy diet etc. I greatly appreciate ANY input!
Hi Steph, I gather you had successful removal of an enlarged PT gland and have a low serum calcium normal PTH, very high urine calcium and high normal serum 1,25d. I am not clear with regard to how long it has been since surgery. Likewise you mention renal health, suggesting your kidney function is reduced. I agree your situation is very complex, and hesitate to say anything about it without more information than would be appropriate on a public forum. I would be willing – if your physicians agree – to review your entire record and offer suggestions to you and your physicians. Of course this would be a courtesy and involve no cost. Please let me know if that would be something desired. Regards, Fred Coe
Hi and thanks for the voice and ear! Complicated initial missed diagnosis of PHPT for >5 years throughout late 20’s and early 30’s despite CA >10.7 PTH 60’s 25 oh d 23-27 , 1,25 d 60’s and hypercalciuria up to 480. Successful surgery revealed intraoperative PTH level of 130 w/ 100% drop but persistent calcium above 10. Calcium dropped to low 9 PTH remained <10 for several weeks. Fast forward to 5 years later, I am seen in ER about twice a year for profound arrhythmia (ER has confused for MI before seeing labs). I have low BP 90/60 days 80/40 while at rest so I am told diuretics are not an option. The hypercalciuria remains in the 400's PTH 20-40 total CA 7.8 -9.4 ionized CA as low as 1.00 regardless of total CA level and without a substantial rise in PTH??Low to low normal phos. Calcitriol remains in 60's and 25 oh d 27. I supplement 3x daily with low dose ca/mag/vit d but cannot predict when hypocalcemic episode will come on. Dr only concerned about low d despite frequent renal issues assumed to be stones passing(interestingly normal gfr- much improved since surgery, went from 60 to 110). At a loss. No idea how to manage, but feel awful and have 9 year old triplets depending on me to be the best mom ever. I am now 43 and a shadow of who I was before that awful parathyroid took over! Also, one of my children has just been diagnosed with hypercalciuria and osteoporosis at age 9. I had PHPT throughout pregnancy(undiagnosed) Any suggestions or thoughts would be greatly appreciated!
Hi Steph, Some of what I wondered about is now here. After five years post PT gland removal you appear to have low serum calcium, normal PTH – but low for the serum calcium! – and very high urine calcium. The entire picture seems like mild hypoparathyroidism, but given treatments you describe matters may be more confused. I have below offered to try to help for real, and hesitate to say much in the absence of complete information. Look at my prior note and decide if you wish to take up my offer. Regards, Fred Coe
Dr.Coe, Its funny, I had inquired about hypo previously and was told it wasn’t worth looking into because it would be highly unusual and not possible because hypo patients have PTH less than 10. I can tell you from a patients perspective that none of this process npr my original hyperparathyroidism was “usual” given extremity of symptoms and relatively low calcium compared to others levels far exceeding mine. I have no words to explain how grateful I am!!! This has been such a long road with so few answers. I would love and greatly appreciate your review. Let me know how to proceed. Thank you sooo much!
Hi Steph, to proceed, assuming your physicians are not adverse, send your records to my secretary Kathleen Dineen. If you email her at kdineen@bsd.uchicago.edu she can make arrangements. I would need all lab data and surgical path reports etc. I rarely do this, but perhaps your case warrants it. Obviously I would be simply advising your physician about possibilities to pursue as mere record review would not constitute the proper practice of medicine. Regards, Fred Coe
Please excuse double post! Though the first deleted and attempted to recap! 🙂
Hi Dr. Coe,
This whole PTH thing is new to me as I’ve only had it tested once just recently while I’m the hospital with left and right flank pain. I’m 32 F with a history of calcium phosphate stones, and just about 6 months ago hospitalized with ischemic colitis, and mild colitis was discovered on my most recent scan. I’ve had two elevated calcium results show in recent blood tests, protein in many recent urinalysis tests showing abnormal. My PTH test came back at 127.7, and my vitamin D test is showing high also at 62.4; yet all doctors I saw in the hospital last week including the Endocrinologist said this is not what’s causing all of my issues (I forgot to mention multiple kidney infections in the last few months, and many other symptoms such as severe fatigue, loss of appetite, high BP, and recently consistent flank pain on both sides). I feel lost as no one is helping or seems to know what’s going on but I am at a loss with all of these issues. Any advice is appreciated!
Hi Nikki, If two fasting blood calcium levels have been high and your serum PTH is high you probably hare PHPT and can be cured by surgery. Fatigue, hypertension, and stones are all common consequences of PHPT. Ischemic colitis is not. Be sure your fasting blood calcium is high on several more occasions and PTH levels in those very same fasting bloods are either normal or high, likewise you need to have 24 hour urine testing to be sure your urine calcium is not remarkably low. Your personal physicians can do this. Of course there may be factors here I do not know about as they are your physicians. Regards, Fred Coe
Hi Dr. Coe,
I’m a 43 year old female. I’ve been struggling with noticeable symptoms for about a year: extreme fatigue, kidney stone (first one in May 18), frequent heart palpitations, hypercalciuria, bone aches.
I have a dr who believes that I have PHPT, but others who disagree. I’m dairy free (since July), I have PTH just outside of the normal range twice and 85 and 89 at other times (it’s been normal several times too). My serum calcium is in the normal range, but my ionized calcium has been high twice. I have moderate pain in the base of my neck and a little to the right on and off for the last year. My kidney function was less after the stone but back to normal now. I’d love to know if you have any thoughts. Thanks so much.
Hi Christine, Low calcium diet is a very bad idea, and I would stop it. Such a diet can raise serum PTH – so called ‘secondary hyperparathyroidism’ but really the expected physiology the body uses to compensate for calcium malnutrition. If on a proper diet your serum calcium is repeatedly above normal, PHPT is not unreasonable. If your serum calcium – always fasting and in the morning! – is not abnormally high you do not have this disease but rather idiopathic hypercalciuria. Nothing you might have benefits from low calcium diet. The ‘normal range’ for serum calcium is tricky as it varies with the analytical platform in the lab in use; in general, high means above 10.1 for morning fasting but labs may post values as high as 10.5 sometimes. Most of the time the lab has no real normals of its own but quotes ranges provided to them. The higher ionized calcium may reflect tighter normal values. I say all this without knowledge of your case so my remarks are for you and for your physicians, the latter are responsible for your care. Regards, Fred Coe
Thanks, Dr. Coe. Just for clarity, the dairy-free diet wasn’t in response to this issue, but a finding of a dairy sensitivity. Yes, my dr stresses always fasting and in the morning blood draws 🙂 Thanks again and please let me know if you have additional thoughts on the dairy-free part. I have benefits from going dairy free that are noticeable so I’m hesitant to re-add and hoping for another route if I need the calcium.
One more clarification – I have high PTH readings from both before and after the dairy-free switch. They’ve been drawing blood for it about 20 times over the last year, trying to “catch” it they’ve told me.
Hi Christine, going without calcium is never a good idea. If you cannot use dairy products perhaps your physicians can devise alternatives for you. If your PTH level is high on or off dairy products there are five alternatives: Even when on them, you had insufficient calcium intake; you do not absorb calcium normally; your vitamin D levels were low when on dairy products – I do not have your data; your kidney function is even a trifle below normal; you really have PHPT. Time and care will tell, they always do. Regards, Fred Coe
Dr. Coe, Thank you again. I have LOTS of data, that’s for sure! If you’d ever be willing to be my doctor (not sure if you do that?) I would be very appreciative of your thoughts/treatment. I’m going to type some various info here to see if I’m complex/interesting enough to pique your interest :). I’m in the Metro Detroit area so not very far but would take some figuring. Info: I was misdiagnosed with Hashimoto’s three years ago where I had many matching symptoms and antibodies present. A year later, still struggling with extreme fatigue, my blood no longer showed those antibodies. Endocrinologist said it was “weird and confusing” that they were no longer present. I have a 1.7mm nodule on my thyroid that we’re monitoring and a cyst on my left kidney. I had racing heart and fainting episode one year ago – all bloodwork (except for PTH and iron) was normal – but I’ve been very aggressively trying to figure this out since then. I have been seeing an MD from U of M who practices holistic medicine and she is adamant that it’s a parathyroid tumor. Endocrinologist from U of M says we need more evidence. After kidney stone, Nephrologist (who I no longer see) advised against Vitamin D supplementation and calcium supplementation so I stopped. Vitamin D was in normal range for a while until I started taking it again (trying for PTH with and without) and after taking D for a few weeks, just had blood drawn again and it’s lower now than before and PTH is 10 points over normal. Calcium (normal and ionized) in normal range. The only other thing is that I’ve been anemic my whole life – even after no more periods. Mentioning that in case there is a pattern with absorption like you mentioned in your response. If you’re not available or interested – no worries at all. Thanks so much again for your time. Happy New Year.
Hi Christine, I am happy to see you personally. Janet Butala (773 702 1475) is our clinic head nurse and can make arrangements. I would need all of your blood and 24 hour urine results, and CD’s of your CTs if any. Bring everything as we cannot trust mailing or promises to send things. You can also email me directly – fcoe@BSD.uchicago.edu – Regards, Fred Coe
Hi, I have elevaterd sodium and alanine aminotransferase. In my urine, calcium oxalate crystals are present and my calcium ionized is high. My PTH , erythrocytes and leukocyte esterase are also high. I have been experiencing lower back and right side pain, confusion, fatigue, nausea and vomiting, stomach pains, my bones ache and I feel depressed for no apparent reason. My doctor is trying to figure out what is wrong with me. She gave me anibiotics for sinus infection and a puffer for my caugh. My symptoms are still present. After my third visit to the doctor, she called me to tell me to drink 2L of water a day, no protein and no salt in my food. Any suggestions?
Hi Maria, alanine aminotransferase in blood, so I presume you have some liver issues. Sodium is in blood and urine, and I presume it is high in your urine as blood sodium in rarely abnormal. If your blood calcium and PTH are high you may well have primary hyperparathyroidism and that can be cured by modern minimally invasive surgery, so I would focus there. Are your bloods always morning and fasting? They need to be for diagnosis. How many have been fasting, morning, and high calcium? Count it up. Is your urine calcium high? Take another look in the article, and be sure of what is found in you vs. what is found in PHPT. Regards, Fred Coe
I have several issues that are probably linked ?
Atrial Fibrillation, Kidney stones and underfunctioning kidneys , underactive thyroid since about 40 years ago ; have had a parathyroid adenoma removed in about 2002 and tend to have high parathyroid levels , but normal calcium levels .also have osteoporosis ; high oxalates and the MTHFR gene .also quite a few food sensitivities .Any suggestions how to manage all this ???
regards
Mary
Hi Mary, I can perhaps do a little. The MTHFR gene variants in adults confer such low risks that NIH recommends against screening for them. Given normal serum calcium levels, I presume the increased serum PTH levels reflect the reduced kidney function, or alternatively low calcium diet or low serum 25 D levels. Given reduced bone mineral levels, you may have idiopathic hypercalciuria as a cause of stones, and it can promote bone mineral loss. High urine oxalate is almost always worsened by low calcium diet, so given the increased PTH and high urine oxalate perhaps you should consider if your diet has sufficient calcium. Supplements are not unreasonable but must be taken with meals to lower urine oxalate. Take a look at the kidney stone diet, as a good place to start. The atrial fibrillation is a separate matter, I think. Regards, Fred Coe
Hello Mr. Coe,
I’m not sure where to post this, hopefully this is ok. I have been searching for help for years, don’t know where to turn. While researching I came across this site. I have been forming kidney stones for almost 20 years at this point. I’ve been to hundreds of doctors and no one can figure out why. I’d also lost weight without trying and cannot gain no matter how much I eat. I have hair loss/thinning as well, both on head and body. Aches and pains are the norm now and I’m only 40. I’ve had doctors suggest parathyroid disease but when labs are run, everything is normal. I know you cannot diagnose, but have you heard of situations of parathyroid adenomas in the presence of normal labs? I am open to any and all suggestions you may have, I’m really suffering. Thank you very much for your time.
Hi Lauren, Given so many stones you must have serum and 24 hour urine testing done, which will surely reveal your problem. PHPT is fasting serum calcium above the upper limit of normal and urine calcium not low and serum PTH level not suppressed. It is an easy diagnosis if blood and urine testing are done properly. But you need full studies as I said including the kinds of stones passed. Here is another article on workup. Take a look and see if you have been evaluated properly. Regards, Fred Coe
Dr Coe: Thank you for sharing so much information on your website. I am writing about my husband, he is 55 years old, 5’6″. 155lb. He had 2 kidney stones in March 2015, 95% calcium oxalate, 5% carbonate apatite. He has had microscopic hematuria for many years, starting before the stones. His urologist said the blood is coming from his kidneys.
He had an episode of kidney/back pain in August, a cat scan was negative for stones, it showed renal cysts.
Our concern now is that his renal function is declining. His eGFR was 62 in August, down from 78 in 2013. His urologist has added a diagnosis of “Chronic kidney disease” .
He had routine fasting blood work on 8/25/19, his calcium was 10.3, vit D 25-OH was 49. He takes vit D 2000 IU daily. Creatinine was 1.29.
Based on the calcium of 10.3, his history of stones, declining renal function and some GI symptoms (GERD, abdominal pain) we asked the urologist to check his PTH. On 9/4/19 his calcium was 9.8, PTH 47. However this was not fasting. I know after reading your articles that this should have been drawn fasting.
Over the past 10 years or so his CO2 level has been high normal or slighty elevated, not sure if that is relevant to his kidney function.
In November 2015 (8 months after his kidney stones) we asked for a 24 hour urine, I will include the results below but I know this needs to be repeated.
Calcium 258 oxalate 11 uric acid 121 citrate 238 pH 5.6 Volume 1L sodium 189 phosphorous 874 magnesium 90
potassium 35 creatinine 1846 sulfate 6 sodium urate 1.2 calcium oxalate 1.42 ss brushite 2.24
Based on the low urine volume he has increased his fluid intake and recently has decreased the sodium in his diet.
We will ask his doctor for a current 24 hour urine. Should we also ask for more fasting calcium/PTH levels? His doctors do not seem concerned about his eGFR because it is still in the normal range, but we would like to figure out what is causing his eGFR to decline and treat the cause if possible. Thank you for any input you may have.
Hi Michelle, The one high serum calcium + stones means what you thought: more fasting serums for calcium and PTH – in the same samples. I always do three, and if all are normal I am done with PHPT as a diagnosis. His urine calcium is high and could cause stones all by itself, but the 24 hour urine concerns me. The oxalate at 11 is very low, low enough I suspect a technical lab failure. Likewise the sulfate of 6. The uric acid of 121 is uninterpretable. If that is the total uric acid in the 24 hour urine it is too low to believe except that some has precipitated during collection. The very low volume and pH of 5.6 makes this a real possibility. Given the creatinine of 1846 I believe it is a reasonably complete collection, and his moderate hypercalciuria and stunning low volume – fluid intake – are more than enough to cause stones and crystals. He is probably crystallizing uric acid all of the time and that can cause hematuria as well as orange – red gravel, if one looks. As for the low GFR, I would worry about blood pressure, and any possibility of impaired urinary drainage. I am sure his physicians are aware of this. You might want to bring some of my suggestions to his physicians for their consideration. They are in charge, and responsible for his care. Regards, Fred Coe
Dr. Coe, Can I get your help. Here are my labs over the course of the last several months. My doctor had me scheduled for surgery to remove Parathyroid, then decided there was a possibility it could be a renal leak. She put me on a dose of HCTZ on Oct. 3, but there is still great confusion – she tells me I am a mystery. Here are my labs and I have done 3 of the 24 urine quest. I am 48 and very healthy and fit. I had 2 occasions of kidneys stones over this past year and have osteopenia. She increased the HCTZ from 1/2 to a whole pill but it was causing my potassium to be low.
PTH Calcium Ionized Calcium
25-Sep High 119.4 9.6 1.26 then started HCTZ
11-Oct 9.8
16-Oct High 108.2 10 High 1.32
21-Oct 75.1 9.8 High 1.30
23-Oct High 115.0 1.27
31-Oct 87.3 9.8 1.26
14-Nov High 116.6 High 10.2 High 1.33
22-Nov High 125.8 9.7 1.26
24 hour Urine Quest
Calcium Urine Quest Bushite Quest Calcium oxalate Quest Sodium Urate Quest
23-Jul High 347 High 11.42 High 6.18 High 6.85
25-Oct High 331 High 9.08 High 2.93 High 4.34
19-Nov Waiting on these results
Hi Sonya, Let’s count your serum calcium levels. Before the thiazide, 9.6; after going on it, 10, 9.8, 9.8, 10.2, 9.8; ionized calcium levels were 1.26 1.32, 1.3, 1.27, 1.26, 1.33, 1.26. The normal range is 1.3 – 1.5 with some variability among labs. I presume all of the bloods were taken fasting and in the morning, as serum calcium rises with meals. If so, the evidence for high serum calcium is weak indeed. The urine calcium values you give are 347 and 331 mg/d, both high. The SS values have nothing to do with the question of hyperparathyroidism or not. As in the article, I do not think your situation is complex intrinsically, but complicated by the drug. PHPT requires serum calcium be elevated and so if I were doing your work I would stop the drug, wait a week or two, and get more fasting morning serum calcium measurements, each with a PTH. If all of the serum calcium values are like most of those you quote I would presume you have mere idiopathic hypercalciuria and a cause of secondary high PTH such as low serum 25D, or low calcium diet, or perhaps even the slightest reduction of renal function. The thiazide raises serum calcium, and so makes it impossible to be sure if your real values are high – I suspect they are not. Please consider all of this as material to discuss with your physician as she is responsible for your care and I am a distant outsider with no real knowledge of your actual medical situation. My remarks, if scientifically correct, may not be pertinent to your case. Regards, Fred Coe
Hello. I have had two uric acid kidney stone surgeries by a urologist. I then went to a parathyroid specialist and she thinks I have hyperparathyrodism and wants to do surgery on my parathyroid glands. My blood calcium levels range from 9 – 10, with my intact PTH of 29. Since I have uric acid kidney stones, is there a connection to these kind of stones and hyperparathyroidism? I’ve not found information on uric acid kidney stones. I have cut way back on animal protein in hopes to help with this stone issue. I also have AFIB, MTHFR, and Osteopenia. I am 66 years old. Your thoughts on my situation would be greatly appreciated. Thank you sir.
Hi Janine, As the article you post on notes, PHPT requires serum calcium be above normal and the values you post are not high. Your PTH is rather on the low side, although that is not a major point. I cannot imagine surgery given a lack of high serum calcium. Uric acid stones occur because of acid urine pH, and can be prevented completely by raising the pH. Of course your physicians are responsible for your care, so you should take my remarks for that they are-comments from a distant outsider. But before surgery they may want to get a second opinion, and that is something I can recommend without much hesitation. Regards, Fred Coe
I had recurrent kidney stones. Then I was diagnosed with hyperparathyroidism. I am fortunate in that I was referred to an endocrinologist (Dr. Garzia Aleppo) that performed tests before scheduling me for surgery, and found I had calcium in my urine and a vitamin D deficiency. Once the vitamin D deficiency was corrected, the parathyroid levels became normal and no more kidney stones.
How do we inform more urologists about the connection between vitamin D and kidney stones? I’ve seen a urologist since that says I must “misunderstand” and vitamin D deficiency does not lead to stones… likewise, I had to fight with my father’s care providers to get them to test his vitamin D levels and try to find a cause for his recurrent stones.
Hi Lynette, I think the sequence of events had another cause. Your high urine calcium may well be idiopathic hypercalciuria. Your serum calcium was presumably normal and your serum PTH high. This latter, the high PTH, was from vitamin D deficiency. The stones arise from the high urine calcium, and vitamin D deficiency is not the cause of that high urine calcium – genetics is the cause. Your father probably has high urine calcium from the same genetic cause, and you inherited it. Vitamin D metabolism is high in genetic hypercalciuria and vitamin D deficiency common. The high urine calcium may well continue to cause stones and needs its own care – this is detailed in the article. Regards, Fred Coe
I am a 69 year old woman. I have been forming stones for over 20 years an have had at least 20 procedures to treat them. About 5 years ago, routine blood tests showed my blood calcium at 10.6. My PTH was not high, but vitamin D was quite low. Sestamibi scan showed a parathyroid adenoma. I had a parathyroidectomy. Just recently I had another BMP and my PTH was 92. I saw my endocrine surgeon and he was pretty sure my parathyroid disease returned. He order another calcium and PTH. PTH came back 47 calcium 9.2. He walked back his diagnosis and said I do not have parathyroid disease. I’ve had a few years of reprieve from kidney stones. Suddenly, I have multiple stones in each kidney and a few weeks ago I ended up in the ED. My calcium was 12.2 at that time. I was treated for the stones and 15 small stones were retrieved. This was only a few weeks ago and I am again having right flank pain. My endocrine surgeon want me back in 6 months. I am not convinced that my parathyroid disease has not returned. Isn’t it unusual for PTH to go from 92 to 47 in such a short time? I know it can fluctuate, but I read the normally the fluctuations are within a few points. Here are my recent stats using parathyroid.com app. 1/2/20: Calcium 10, PTH 92, 1/20/20: Calcium 12, 1/24/20 Calcium 9.2, PTH 47, 2/9/20: Calcium 10.2, 2/11/20: Calcium 10. The app analysis says that is is “very likely” I have parathyroid disease, if the app is indeed reliable. During the 20 years of forming stones, my calcium was under 10. Vit. D and PTH never checked.
Hi Angela, Your serum calcium levels do indeed seem high but they vary a lot. Here is what I would do: Get morning, fasting, serum calcium levels, fasting since midnight before the blood draw, and find out what your serum calcium really is. 12 is very high, and so is 10.2. PTH is regulated by serum calcium, so you cannot interpret a PTH without a serum calcium to use it with. Sometimes in early recurrent PHPT serum PTH is high fasting and serum calcium normal or high normal, but with food serum calcium rises abnormally and PTH falls into the normal range – the calcium is regulating PTH but at an abnormal way. ANyway, the diagnosis depends on fasting morning serum calcium values which will gradually rise into the abnormally high range if you have recurrent disease. You do not mention urine calcium, but often 24 hour urine calcium rises a lot as PHPT recurs. I suspect it has, and I would get a lot of serum calcium measurements. Regards, Fred Coe
Dr Coe, how many days or weeks apart should you retest the blood calcium and PTH to check for Primary Hyperparathyroidism, you mentioned morning fasting and to do at least 3 times. Thank you!
Hi Carol, I tend to get bloods to the convenience of the patient. What matters is fasting, morning blood draw, and calcium and PTH in the same sample. If calcium levels are not entirely within the normal range I keep repeating until it becomes obvious if the average is really high – if so we have a probability of PHPT. Regards, Fred Coe
Dr. Coe, I was sent to a general surgeon by my primary doctor for a third opinion on primary hyperparathyroidism because of forming stones and my labs being iffy with a couple higher PTH’s but only one high calcium. My endocrinologist sent me to Quest lab to repeat and they were normal and she does not believe I have this. I was dismayed that the surgeon who seems very knowledgeable and told me he does these surgeries often and spent a great amount of time with me told me it did not matter time of day or fasting that I had the PTH and Calcium blood tests done, he said over and over it didn’t matter if you were not fasting which my last two were not because the endocrinologist and urologist did not tell me to do them fasting. I stated to the surgeon after doing research online that they must be done fasting and in the morning, he just kept saying that does not matter. Of note, he also leans toward the endo doc that I do not have this and to get more labs to confirm for sure so that was encouraging that he wasn’t out to just do a surgery. I just wonder why so many physicians that should have knowledge when dealing with this illness don’t see how important this is. Do you come across these same type of physicians that don’t realize these need to be done fasting? I can possibly understand a primary doctor not knowing since it’s not their specialty but these other docs should know. I thank you for telling me this because I will make sure I get them fasting at my next visit.
Hi Carol, I hope I am not making problems for others here. Fasting does matter, as serum calcium rises with meals. However if your serum calcium values are all normal (I usually require less than 10.1 ml/dl to be satisfied about normal, whatever the lab normal range), then fasting does not matter as serum calcium would go up with meals and is still not high. Just be sure your serum calcium values are below 10.1. For PTH, it goes down with meals, because serum calcium rises. This is all normal physiology, and one can find these very small changes in our publications and others. High PTH with normal serum calcium can be low calcium diet, low vitamin D levels, or mild reduction of renal function. It is not treated surgically. Of course PHPT almost always includes high urine calcium excretion absent kidney disease, or some other complication. The crucial element is serum calcium, and I am afraid I have to disagree with any and all who do not believe in standardizing time of day and fasting to compare results to normal people. Regards, Fred Coe
Dr Coe, If a person truly has PHPT and they have surgery to remove the abnormal gland or glands and that cured their Calcium Phosphate stones would that also lower the urine PH in someone with high urine PH or is that a totally separate issue not connected to PHPT?
Hi Diane, PHPT is a disease of parathyroid hormone excess and cure is not only stones but bone disease, hypertension, ulcer and pancreatitis risk, and more. Calcium phosphate stones by themselves are a special problem and need a different kind of care. You cannot lower urine pH in CaP stone formers, at least I have not found a way, but you can work around to stop the stones. Regards, Fred Coe
Dear Dr. Coe, Thank you for the informative article. I had to laugh at the statement regarding Hyperparathyroidism Jaw Tumor Syndrome: “this disease is so colorful one diagnoses it readily and cannot confuse it with commonplace PHPT…” I had my first symptoms, including many, many kidney stones as a teenager and for 10 years until I was diagnosed with PHPT and had my first surgery at 27 and was cured. Until I had it again when I was 48 and had a second surgery and was cured. And then had PHPT again and another surgery at 58 and was cured. I’m down to just a bit of 1 parathyroid. None of my endocrinologists and only 1 of my surgeons ever mentioned a potential genetic cause and none would order genetic testing. I finally ordered my own genetic testing at 60 and got a result of HPT-JT. Hopefully it will be more readily diagnosed for others! Thanks again.
Hi Maryce, Thank you so much! My article does not include the CDC73 gene abnormality but should. It is a rare disease. You were wise to go ahead on your own. I presume cinacalcet is being considered if your serum calcium is high. But that will increase urine calcium. The alternative is to remove the remaining PT gland, and that has its own problems. I hope you are being cared for in a major university center. I should mention that Mayo has a rare disease program. Regards, Fred
Dear Dr. Coe,
I had PHPT for 10 years before it was diagnosed. I had a parathyroidectomy, total thyroidectomy, PCNL, EWSL, Bilateral Ureteroscopy, and two fractures. I had recent labs reflecting blood, calcium oxalate crystals, WCB, RBC, and protein in my urinalysis along with flank pain recently. My doctor said years ago my kidney tissues have calcified (similar to Randall’s plaque) and there is nothing more he can do. I had a recent CT which reflects I have innumberable kidney stones – 6 and 7mm being the largest in the lower and mid pole. To my understanding these stones are not in a favourable location to pass. My doctor does know why I am having flank pain with non-obstructing stones. Please let me know what imaging/tests I can request to see if I have IMCD and BD plugs or any other information. Thank you for your incredible information and website.
Hi Susan, Have you continued to form stones despite cure of your PHPT? Or are all of the stones relics of years past? Plaque certainly forms in PHPT and can foster stones. But what are your current 24 hour urine stone risks? Is calcium high? You cannot image plugs or plaques, I am sorry to say. We did all of our work using intra-operative imaging. Get re-tested for stone risks or if you have been look closely for clues to continued risk. Regards, Fred Coe
Hi Dr Coe,
I am wondering what you think about my levels
I am 51
Thyroid nodule TrADS 4 biopsy results indeterminate thyroseq genetic testing negative 3% cancer risk
Small homogeneously hypoechoic oblong nodule inferior to the right thyroid lobe measuring up to 1.0 cm, may represent lymph node or parathyroid adenoma, though lack of significant vascularity favors the former. 4d CT scan unable to determine exact nature due to artifact from recent spinal fusion hardware.
Initial labs PTH 85 calcium 9.8 ionized calcium 1.35 vitamin d 19
After vitamin d replacement
Vitamin D 31 ;pth 81 calcium 10
24 hour urine 0.70g/24h
Repeat 24 hour urine 0.51g/24h
I have been reading a lot of your articles and would appreciate your input
Melissa
Hi Melissa, I think you have secondary hyperparathyroidism from prior low vitamin D levels and (pure guess on my part) low calcium intake over a long time. Your urine calcium is very high if you really mean 0.5 – 0.7 grams (500 to 700 mg) of daily calcium loss. That is so high as to raise eyebrows and suggests underlying diseases of bone or perhaps genetic. Your physicians really need to determine what is the cause. One caveat: your evaluation is not adequate to me. I would get several morning, fasting bloods for calcium and PTH together and be sure the serum calcium is indeed below 10 on both occasions as if it is as high as 10 one must suspect PHPT as in this article. Regards, Fred Coe