 I do not know why anyone would diagnose distal RTA (dRTA) very often. As I will show you it has colorful and unusual characteristics as unmistakable as rare, so diagnosis is not difficult. But many more people think they have than have it. In my 50 years of kidney stone prevention I have perhaps a few dozen examples or so, out of many thousands of stone formers.
I do not know why anyone would diagnose distal RTA (dRTA) very often. As I will show you it has colorful and unusual characteristics as unmistakable as rare, so diagnosis is not difficult. But many more people think they have than have it. In my 50 years of kidney stone prevention I have perhaps a few dozen examples or so, out of many thousands of stone formers.
This is another of those long, elaborate articles only the most devoted read.
Even so, elaborate as it is, this article tells only part of the story. It simplifies or simply ignores the mechanism for low potassium in dRTA, and left for another time its genetic causes, and also the bone and mineral disorders and treatment outcomes. I forgive myself, as just this part has been most taxing to write and is equally so to read.
In a subsequent article I hope to expand on diagnosis and treatment, the bone and mineral disorders, genetic transporter disorders, and take up the novel modern issue of acid retention and its effects on kidneys. So consider the present article a part of my planned contribution.
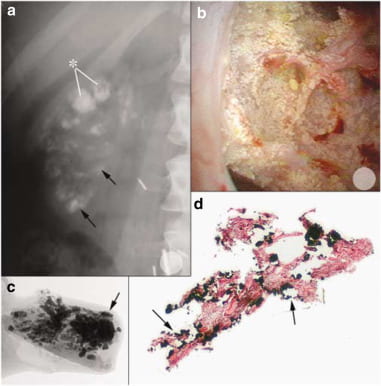
The featured illustration of kidney tissue from a patient with dRTA shows many crystal deposits on a radiograph (panel a), that at surgery mostly are calcified deposits (panel b) that nearly replace papillary tissue (panels c and d).
What is distal RTA?
Kidneys make urine more acid than blood because most of us eat a diet that imposes an acid load on the body and kidneys need to remove that acid. But apart from balancing acid excretion to diet acid load, unless kidneys acidify urine calcium phosphate crystals may form in such profusion as to block kidney tubules and produce kidney stones.
This happens because as they conserve water kidneys concentrate urine calcium phosphate salts far above their levels in blood. If they simultaneously make urine more acid than blood the calcium and phosphate will stay in solution and make no crystals. That is the normal state of affairs.
But what would you predict might happen if by magic – bad magic – kidneys lost their normal ability to make urine acidic? Perhaps not all their ability, but some significant part of it? Yet withal, retained enough of their life sustaining glomerular filtration so that their other functions were close to or in fact normal.
You would predict acid would accumulate in the body and cause trouble, perhaps leach mineral from bone. Likewise you would predict calcium phosphate crystals will plug tubules and make stones. This because preservation of filtration assures delivery of ample calcium into the late nephron and final urine.
You would be right both times.
That is the very make and mark of distal renal tubular acidosis (dRTA).
Bad magic.
What Does ‘Distal’ Mean?
The nephron begins at the glomerulus. The proximal tubule lies just beyond it. Prior articles do a reasonable job of showing where things are. Tubule cells acidify the filtrate in the proximal tubule, and then again later on, in the collecting ducts. When the latter is defective, urine itself is invariably too alkaline predisposing to calcium phosphate crystals and plugs. Such an alkaline urine is common among idiopathic calcium phosphate stone formers who certainly do not have dRTA. In those who do have such a defect, stones and crystals are usually far more massive and damaging to kidneys.
When the former is defective (proximal RTA), filtered bicarbonate cannot be fully reabsorbed as in normals, so more is delivered downstream to the collecting ducts. There, the cells are competent to secrete acid, but the amounts of bicarbonate can be so high as to use up all of it and escape into the urine. The result is too alkaline a urine. Stones, however, are not so likely. Serum and therefore filtrate bicarbonate falls, better matching what remains of proximal reabsorption so the distal delivery slows to a trickle.
But, sometimes a ‘proximal’ problem can indeed cause stones. For example drugs like topiramate reduce proximal tubule acid secretion and lead to calcium phosphate stones. In part this may reflect intermittent dosing, so serum bicarbonate rises between doses, and partly a more acid blood that lowers urine citrate, a powerful anti – stone molecule.
Our concern here is the distal form of RTA.
Clinical Laboratory Appearance
Should we not expect to see high urine pH – alkaline urine – with acid retention in the blood?
Some Explanation of Terms
pH
Proton concentration varies over millions of fold from acids to alkali, and being a logarithm, pH spans that range gracefully. In fact, pH is the log to the base 10 of 1/[H+] where [H+] is the concentration of hydrogen ions – protons. For example, water has a pH of 7, meaning the concentration of protons is 10-7 (0.000,000,7) moles/liter. Blood is nearly the same, pH 7.4. But urine is normally pH 6, more than 10 times more acid than blood.
Blood is held at a pH just above that of water by a buffer system consisting of bicarbonate and carbon dioxide (CO2) gas. The latter is regulated by the brain using the lungs to clear CO2 from our metabolism at just the rate desired to maintain blood pH. Bicarbonate is a negative ion that can take up a proton – acid radical – or give one off. So it is a ‘buffer’ – meaning by its donation or uptake of protons it can stabilize the pH of blood.
Bicarbonate
When food and metabolism add protons to blood, those protons are ‘buffered’ by blood bicarbonate. But that buffering has a strange property. As it takes up a proton, bicarbonate becomes carbonic acid (H2CO3) that almost immediately decomposes into dissolved CO2 (dCO2) and water:
H+ + HCO3– <—–>H2CO3 <—–> dCO2 + H2O
Dissolved CO2 (dCO2) is in equilibrium with CO2 gas whose pressure in the blood (pCO2) is fixed by the brain and lungs. The lungs carry away any excess, so the partial pressure of CO2 gas in our blood can be very constant. Given this, when a bicarbonate buffers a proton It disappears into thin air – CO2 gas, actually.
The arrows point both ways, because these equilibria can move back and forth. If kidneys remove a proton – they do this to make up for what we eat – things simply reverse. Just imagine a tiny tweezers removes the H+ at the left of the equation above. Immediately, the lost proton is replaced by a proton from H2CO3, that then becomes HCO3–and the ‘lost’ H2CO3 is replaced from dCO2 and H2O.
Does this mean that new bicarbonate appears in blood out of thin air?
Yes. As kidneys remove acid to make up for our diet, they make new bicarbonate. They make it in the capillaries around the renal tubules that secrete acid into the final urine, and the bicarbonate flows into the renal veins, and thence into the general circulation through the vena cava.
Does that mean if we anchored a tiny canoe in the vena cava just at the mouth of a renal vein, and dipped a pH meter into the blood around, us the pH would be higher at the vein’s mouth than in the central parts of that great vein?
Yes.
Total CO2
What we measure in blood is the total of HCO3– + H2CO3 + dissolved CO2 so we call it total CO2 or just TCO2.Of these, HCO3– massively predominated. So when TCO2 goes down, acid has been retained. When it goes up, acid has been removed – or new alkali added.
Lets Use the Terms
The big graph shows serum TCO2 and urine pH. For simplicity I have left out the ‘T’.
Common Stone Formers
The tiny blue dots are my data from common calcium stone formers. Most of the points center between 23 and 30 for total CO2. Urine 24 hour pH averages about 5.9 but ranges high and low because these people have normal kidneys that are adjusting for the diet they have chosen to eat.
Normal Men and Women
Blue large circles are from normal people we have studied. They gave us 24 hour urines and let us draw a blood. Nothing more. Remarkably, their points overlap exactly with those from the common stone formers.

dRTA and Acid Loaded Normals
The red circles are clinical measurements from my patients with known dRTA. A few could not or would not discontinue their alkali treatments; their points show normal serum CO2 values and very high urine pH.
The red up triangles show dRTA cases given an acid load to force urine pH as low as it can get. I have plotted this low pH against their serum CO2 values before the acid load – the value one would see clinically. So low values mean the patients have abnormal blood acidity even without an acid load.
The blue up triangles, from our own and other published studies, show acid loaded normal people. Once again, when possible, I have plotted their lowest urine pH values – from acid loading – against their CO2 values before the acid load. The big blue square is an odd patient from one study – see below.
For those who want to see the data I have collected from published sources, here is a link to the references and the values plotted on this figure. My clinical data are not in the spreadsheet because never published explicitly in this form.
What Does the Graph Say?
Normals and Common Stone Formers
The blue circles and tiny dots show us the variable urine pH of normal people and common stone formers. Their serum CO2 values are almost all to the right – higher than – those for the red symbols (dRTA) on average.
Acid Loaded Normals
Their most acid urine pH values are all below 5.3. A few cases lacked pre-acid load serum CO2 so I plotted the values after acid loading. The one blue square shows a single patient labeled as ‘incomplete’ RTA in one of the references. Although urine pH fell normally, a more elaborate test failed – I view the person as having normal renal acidification.
dRTA
When the kidneys are disabled by dRTA, either genetic or from special kinds of kidney diseases, the serum CO2 falls because acid accumulates. It accumulates not because we gave an acid load but because the kidneys cannot cope with our normal acid load from food. But the lower serum CO2 – meaning acid blood – cannot drive kidneys to lower urine pH. So most of the red up triangles have the unique signature of dRTA: high urine pH and low serum CO2.
Incomplete dRTA
What about the red triangles with normal (above 23 mmol/l) serum CO2? For them I show their lowest urine pH after an acid challenge. It does not reach below the limits for normal people. These cases are labeled ‘Incomplete RTA’ and I accept them as they cannot lower their urine pH as much as normal people would given an acid load (blue up triangles). Unlike the dRTA patients with reduced serum CO2 those with incomplete dRTA can stave off ‘acidosis’, an acid blood as gauged by reduced serum CO2. But their kidneys respond to an acid load challenge less well than normal thus disclosing some defect in lowering urine pH.
Do Patients with dRTA Make Stones?
Yes. The cases, my few and those in the papers I selected, mostly came for care because of stones. Some had bone disease, some children did not grow properly. These are thought to be consequences of the acid retention.
They not only make stones, but generally the stones we expect: Calcium phosphate, the stones that form in alkaline urine. Like calcium phosphate stone formers, patients with dRTA can form mixed stones that contain some calcium oxalate, and occasionally produce stones predominantly of calcium oxalate. But in general calcium phosphate predominates. Likewise, patients with dRTA and idiopathic calcium phosphate stones plug terminal portions of their nephrons with masses of calcium phosphate crystals.
But unlike the not uncommon idiopathic calcium phosphate stone formers, those with dRTA have unrelentingly high urine pH and acid retention. Perhaps because the nephrons have lost their ability to lower pH, so tubule fluid is perpetually alkaline, they form much larger masses of crystals, in the urinary system and as terminal collecting duct plugs. Radiologists often encounter massive calcium deposits in the kidneys and label the condition ‘nephrocalcinosis‘,
Tissue Findings
We were not the first group to study the kidney tissue in dRTA, but those before us sampled the kidney cortex, where glomeruli and proximal tubules predominate. We expect crystals to form not there but in the terminal portions of the nephrons, where urine is most concentrated, and it is precisely from there we obtained our tissue samples, and within that narrow precinct where we made our observations.

Radiographs Can Mislead
Among our few cases (5) we described two with multiple masses of calcifications in the kidneys – nephrocalcinosis. As in phosphate stone formers, these calcifications can be stones or masses of tissue calcium deposits.
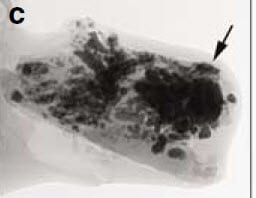
Here is one kidney. Many calcified objects more or less fill it. Arrows point to some obvious clusters. This is a perfect image of what radiologists correctly label ‘nephrocalcinosis’. It is the name I would use. The large clusters marked by asterisks were in an obstructed upper pole calyx and certainly masses of crystal, but whether in tissue or the collecting system we would not be able to find out.
Inside the kidney, at surgery, no stones. Instead, the papillae were filled with crystals inside the tubules.
Here is a biopsy from a papilla. We studied it using high resolution micro-CT. Inside the tissue are masses of crystal deposits filling much of the space. In fact, when cut by a microtome to make microscopic sections, it shredded as the knife could not well cut the crystal deposits and pulled the tissue apart. The arrow points to a huge agglomeration of crystals in tubules.
Kidneys in dRTA
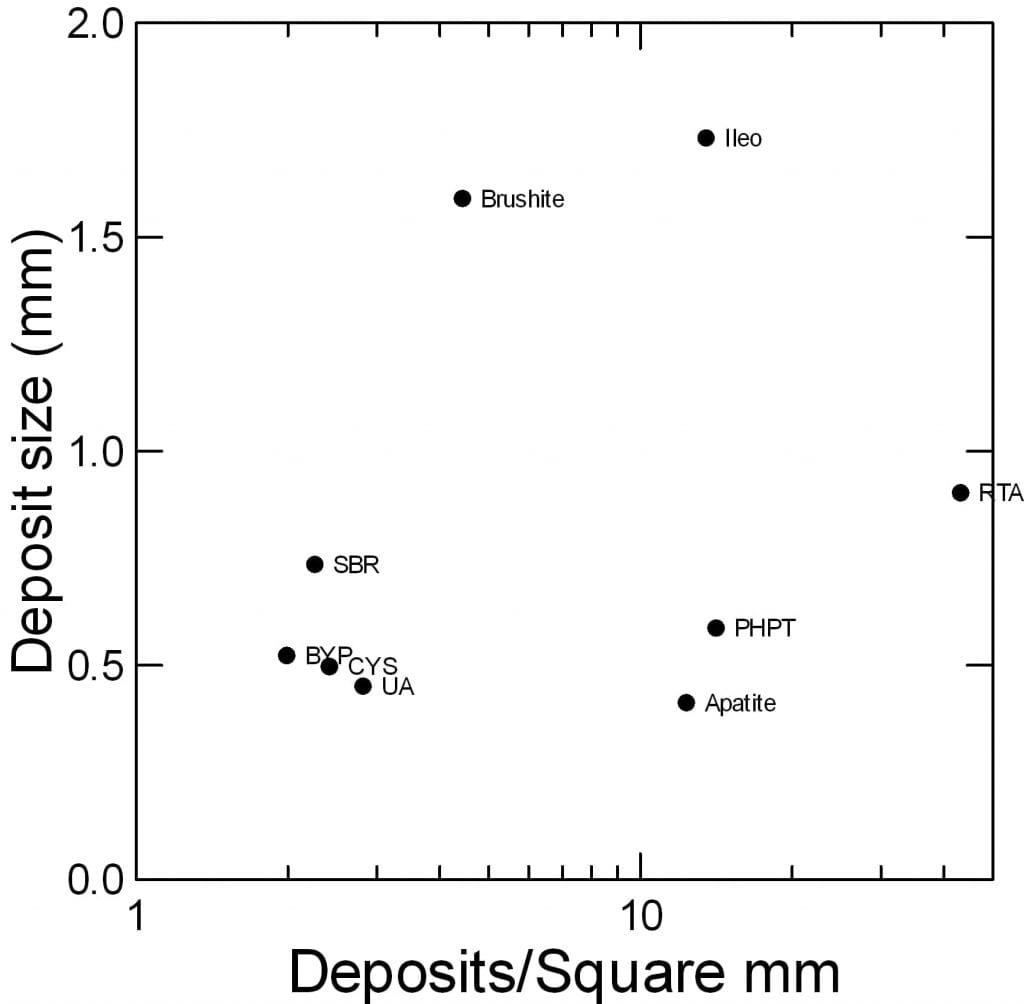 You might say that in plugging their terminal nephrons with crystals, and forming mainly calcium phosphate stones, patients with dRTA are just like calcium phosphate stone formers, who do the very same thing. But saying this is to miss the main point. When compared quantitatively in terms of numbers of deposits and deposit size dRTA vastly outweighs other stone diseases.
You might say that in plugging their terminal nephrons with crystals, and forming mainly calcium phosphate stones, patients with dRTA are just like calcium phosphate stone formers, who do the very same thing. But saying this is to miss the main point. When compared quantitatively in terms of numbers of deposits and deposit size dRTA vastly outweighs other stone diseases.
Elsewhere we have compared deposit size and numbers in multiple stone diseases, but not exactly as I propose to do here.
Idiopathic Calcium Phosphate Stone Formers
Brushite stone formers – one of the two types we encounter – plug occasional tubules with large masses of crystals, so that one tubule is utterly destroyed, but most tubules are fine. See its point on the graph? About 3 deposits per mm2 but over 1.5 mm in size.
Apatite stone formers plug a higher fraction of tubules but with much smaller deposits. Their point on the graph shows about 12 deposits per mm2 but only about 0.6 mm in size.
dRTA
By contrast, dRTA is a more diffuse disease, affecting more or less all of the tubules. So mineral deposits are much more dense – about 45 deposits per mm2. They are also larger than apatite deposits, about 0.8 mm, so the total volume of tissue occupied is much greater.
Other Kinds of Stone formers
Notably, primary hyperparathyroidism (PHPT) is about like apatite stones and ileostomy – the latter a state of constant high risk from dehydration.
The other stone diseases cluster to the far left: small bowel resection, obesity bypass, cystinuria, uric acid stones. They cause few and small deposits. The most common calcium stone disease of all, idiopathic calcium oxalate stones, causes very few and small deposits. We found almost none; the Mayo Clinic group described tiny ones.
None match dRTA for its sheer diffuseness, its contamination of so large a fraction of tubules with crystals. Take another look at the micro-CT. You can see very little uninvolved tissue.
What Makes Crystals So Massive?
For this analysis, I will use only dRTA cases in my clinical series because their lab values were obtained exactly as were values for the other stone forming groups I need as a contrast.
Urine pH
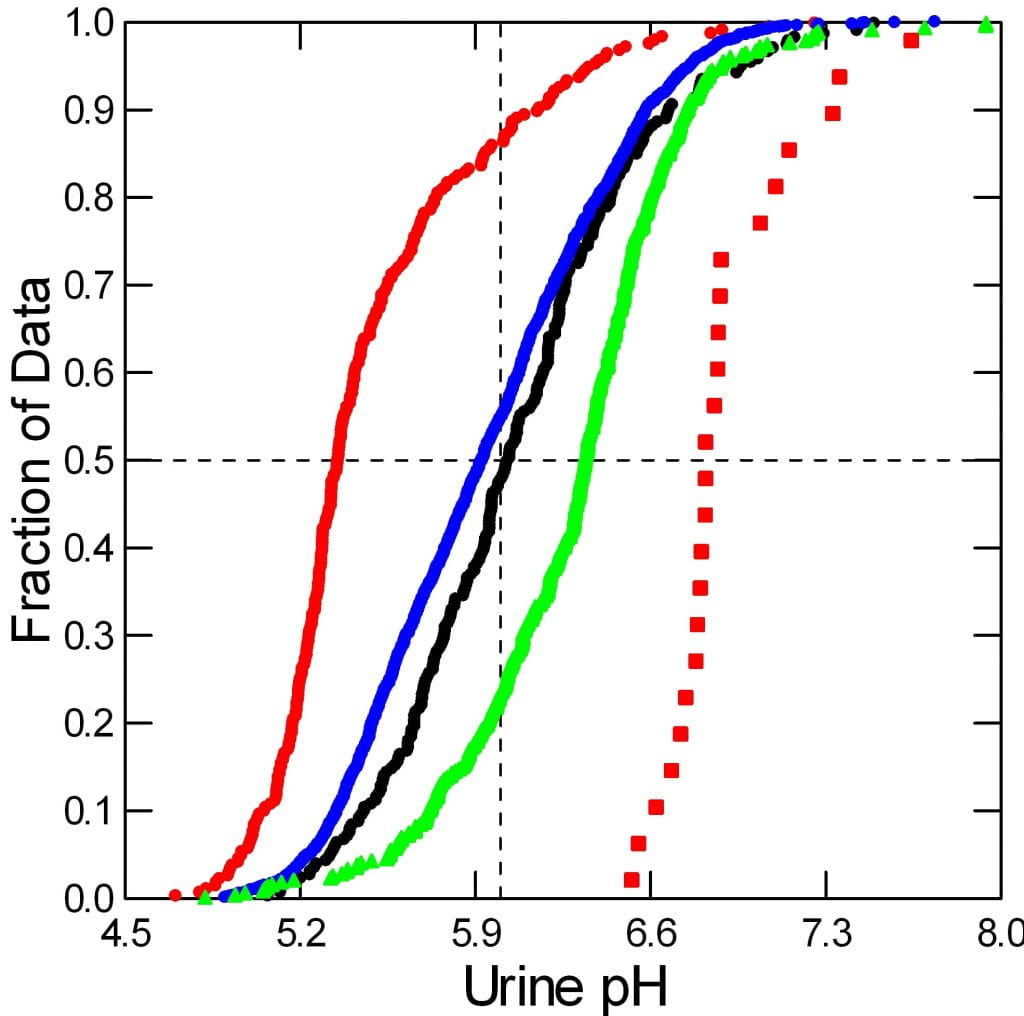
Certainly the high urine pH is part of the story.
This quantile plot (qplot) shows the distribution of values for uric acid stone formers (red circles, to the left), CaOx stone formers (blue circles), normal people (black circles), CaP stone formers (green triangles), and our dRTA patients in red large squares.
It is true that the CaP stone formers make a more alkaline urine than do the CaOx stone formers, and that is a perfectly reasonable mechanism for their stones. The higher pH removes a proton from the second site on phosphate so a higher fraction of urine phosphate has two negative sites to mate better with calcium that has 2 positive sites – I know this is a bit oversimplified, but it is not wrong.
Likewise, the much lower urine pH of uric acid stone formers is indeed exactly why they produce those self same stones.
But RTA is a powerful exaggeration of high pH. The whole group of patients is shifted about 1/2 pH urine up, and pH is a logarithm. This will free up even a higher fraction of phosphate to mate with calcium, thereby raising supersaturation for any urine calcium, volume and citrate.
Urine Calcium and Citrate
dRTA causes not only remarkably high urine pH, but also an extreme deficit of urine citrate, and the latter certainly contributes to the exuberance of crystallizations.
The Calcium to Citrate Proportion
Other articles explore the remarkable properties of urine citrate. It binds urine calcium into a highly soluble complex so it is not free to combine with oxalate or phosphate. If crystals do form, citrate can attack their lattice and abridge growth, even in some cases destroy them.
Essentially, the citrate in urine competes against oxalate and phosphate for the attentions of calcium. Especially when pH is high, and a large fraction of phosphate has two negative charges to offer as calcium binding sites, the relative concentrations of calcium, phosphate and citrate are critical in determining how much calcium will combine with each. Moreover, to poison new formed crystals and abridge their growth, citrate must be free – not bound to calcium. So howsoever wonderful citrate may be as a calcium binder and crystal inhibitor, when it is outweighed by excess calcium its magical properties do not so much wane as become inadequate to the tasks.
Calcium to Citrate Plot
This means that the relationship between urine citrate and calcium cannot be but critical in the assessment of stone forming conditions.
The best way to visualize this relationship is simply to graph one against the other in comparable units. On the graph below, urine citrate is plotted against urine calcium, both in mmol/d. The long blue dashed diagonal line of identity bisects the graph. Points above the line mean an excess of citrate over calcium, below the opposite.
Uric Acid Stones
Uric acid stone formers (red circles) scatter above and below the line of identity. The best fit regression line – red line – runs below identity. Note the regression lines stretch no further to the right or left than the highest to lowest urine calcium values.

Normals and CaOx Stone Formers
Normals
Like uric acid stone formers, values from normals scatter above and below the line of identity (black triangles). The black regression line is not different from uric acid.
CaOx Stone Formers
Being so numerous, I plotted calcium oxalate stone formers using tiny blue dots. Otherwise they would obliterate the other symbols.
Their regression line (solid blue line) stretches to the right border of the figure because of hypercalciuria – that much raises urine calcium. Although the regression lines for uric acid stone formers, normals, and CaOx stone formers are more or less the same, one can see the last of the three is below the other two. The lower proportion of citrate to calcium would favor calcium crystallization. But the lower pH would make that crystal CaOx, not CaP. And in general, the result is stones, with little or no tubule plugging.
CaP Stone Formers
These nearest relatives to RTA have, like them, a higher than average urine pH and, interestingly, a lower level of citrate in relation to urine calcium than the other aforementioned groups. See where the CaP stone former points (green) have a regression line that moves in parallel with the blue line for CaOx stone formers but lies lower down. For any amount of calcium they have less citrate to put against it. And, they have a higher urine pH. So they would be facile producers of calcium phosphate crystals, and plug some tubules, like dRTA patients do. Their regression line runs to the right hand margin of the graph because CaP stone formers, like CaOx stone formers, are often very hypercalciuric.
RTA
At the very bottom of the graph, in large red squares, they have more or less no citrate at all to balance their calcium. Their regression line is flat and also short, the latter meaning they have much less urine calcium losses than either CaOx or CaP stone formers. But despite that limitation, their exteme citrate deficit and extreme pH elevation permits formation of masses of crystals to fill most collecting ducts and create numerous calcium phosphate stones.
Statistics
Adjusted Mean Citrate Excretions
One cannot leave this without some quantification of differences. In a general linear model with urine citrate as dependent, urine calcium as independent and stone type as categorical variable, the mean urine citrate excretions (means in mmol/d) adjusted for urine calcium differ.
| Factor | Level | Mean | Standard Error | N |
| STNTYP$ | CAOX | 2.8018 | 0.0343 | 1,770.0000 |
| STNTYP$ | CAP | 2.3260 | 0.0733 | 413.0000 |
| STNTYP$ | NONE | 3.1632 | 0.1073 | 246.0000 |
| STNTYP$ | RTA | 1.0854 | 0.2953 | 24.0000 |
The highest citrate is among normals, the very lowest among dRTA. In a simple comparison between all groups taken two at a time, differences are highly significant – p<0.0001 in all cases except CaOx vs. None (p=0.0073) and CaP vs dRTA (p=0.0003). These p values are adjusted for multiple comparisons. The high significance for dRTA is remarkable given I use only my small group of cases. I chose against merging data from published papers with mine, because their conditions of lab measurement and patient condition might not match.
Slope Dependence of Urine Citrate on Urine Calcium
Although the slopes look different on the graph – citrate rises less with calcium in dRTA, for example, this difference is not significant – p=0.26. The four regression slopes are: CaOx SF = 0.197, CaP SF = 0.171, Normals = 0.156, and dRTA = 0.027 mmol/d citrate per mmol/d calcium. Given the huge spread, I suspect with more data the low slope for dRTA would be highly significant.
You might ask why urine citrate indeed varies so remarkably with urine calcium. In our regression, the F value for urine calcium as a covariate was 17, that for stone former type was 6.28. I do not know why.
What Makes Urine Citrate So Low?
Kidneys can let filtered citrate out into the urine, or reabsorb it into its proximal tubule cells where it becomes fuel for energy production. This reabsorption is regulated by the acid – base status of the blood and the kidney tubule cells.
Acid Retention
The ‘acidosis’ of dRTA is reflected in low serum CO2. Kidney cells can sense that condition and reabsorb citrate.
One might at this point like some hard statistics about the actual values of serum CO2 even though the large pH graph has made a visual impression.
Serum CO2 is low meaning blood is acidic. In an ANOVA without any adjustments (values for least square (LS) means are mmol/l):
| Factor | Level | LS Mean | Standard Error | N |
| STNTYP$ | CAOX | 26.914 | 0.058 | 1,722.000 |
| STNTYP$ | CAP | 26.477 | 0.120 | 399.000 |
| STNTYP$ | NONE | 26.509 | 0.286 | 70.000 |
| STNTYP$ | RTA | 21.375 | 0.488 | 24.000 |
Serum CO2 is obviously low in dRTA, the very hallmark of the disease. Pairwise comparisons show dRTA is below each of the other groups with very high significance (p<0.0001 all three comparisons with full adjustment for multiple comparisons).
Blood Potassium Is Often Low
 Not only does dRTA cause acidosis, it causes potassium depletion. In turn, potassium depletion lowers the pH inside kidney cells, and thereby and independently of acid retention, raises citrate reabsorption so urine citrate falls.
Not only does dRTA cause acidosis, it causes potassium depletion. In turn, potassium depletion lowers the pH inside kidney cells, and thereby and independently of acid retention, raises citrate reabsorption so urine citrate falls.
Normal Subjects
Normal subjects and a mass of calcium stone formers are shown as large blue circles and a cloud of tiny blue points. The normal points overlay those from stone formers because common stone formers, before treatments that might alter it, have normal values for serum potassium and CO2. The large blue square is an ‘Incomplete’ RTA patient, the large blue triangle a normal mean, both from published studies.
dRTA Patients
Values from dRTA patients are in red up triangles. The larger red triangles are from my practice, the smaller ones are published data. The spreadsheet of all published data is available for review.
Not always, but very often, dRTA produces a low serum potassium. Because most of the potassium in the body is inside cells, low serum values almost always mean low potassium levels inside cells.
Some Statistics
Although serum potassium and CO2 appear correlated, they are not (Using only my own clinical data, p for correlation in a general linear model = 0.166). But in a pairwise comparison of mean values adjusted for serum CO2 the mean value of serum potassium from dRTA differed from those of normals, and CaP and CaOx stone formers: dRTA=3.73 vs. normals = 4.18, CaP SF = 4.24, and CaOx SF = 4.29 mEq/l, respectively, p< 0.0001 for all comparisons but dRTA vs. none (p=0.003).
Although I hesitate to make direct comparisons or results from our clinic and the published sources in my spreadsheet, the latter give results much like ours. The mean of 41 published serum potassium values from dRTA cases is 3.64 (95% CI for mean 3.48 – 3.81). Therefore the mean of our cases – 3.73 lies within the 95% CI for the published cases.
The Anion Gap
As though I have trapped you in an inexhaustible maze, yet another detail of dRTA demands our attention. So esoteric a point yet one we must encounter and wrestle with.
Diet Acid Load
 Protons do not come into the blood unattended. They come with a negative partner, the so called proton donor. Carbonic acid is a proton donor – it can give off a proton. But the one I care about – and you need to care about – is sulfate. The diet acid load is mainly – for the most part – sulfuric acid derived from the oxidation of sulfur on the two amino acids cysteine and methionine. Both occur naturally in food.
Protons do not come into the blood unattended. They come with a negative partner, the so called proton donor. Carbonic acid is a proton donor – it can give off a proton. But the one I care about – and you need to care about – is sulfate. The diet acid load is mainly – for the most part – sulfuric acid derived from the oxidation of sulfur on the two amino acids cysteine and methionine. Both occur naturally in food.
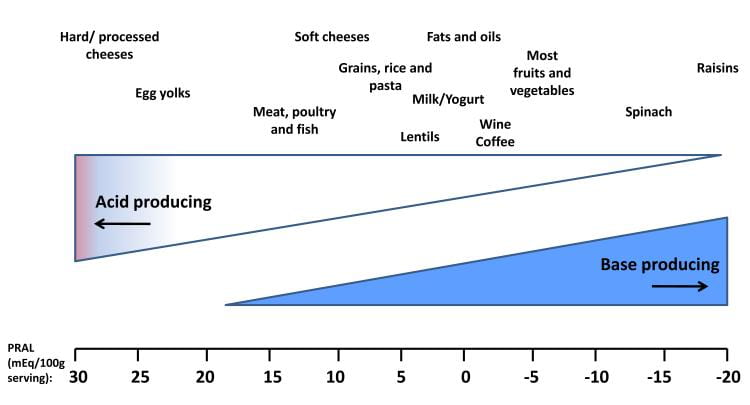
Offsetting the sulfate acid load, foods contain considerable amounts of mostly potassium anion salts that are metabolized to alkali – bicarbonate – in the body. The difference between the sulfate and alkali anions measures the acid load from a given food, and the average of foods the acid load from a diet.
Cheeses and egg yolks are highly acid loading, raisins alkali loading, as examples. One can find charts depicting a majority of foods. Here in the US and most of the West, the diet is overbalanced toward net acid load. If it were biased toward alkali load, dRTA would not appear as it does. Tubules would still be unable to lower urine pH but serum CO2 need be no lower than among normals. Bone mineral could remain stable, and urine calcium lower. It would be as if dRTA were treated with extra potassium alkali.
Reduced Kidney Function
As kidneys succumb to disease, they lose their ability to remove diet acid efficiently, and serum CO2 falls, just like in dRTA. But, the anions, sulfate mainly, from which the extra protons arose, are retained along with the protons. This is because kidneys have lost their normal glomerular filtration rate, and cannot clear sulfate from blood normally. The disease may or may not have impaired the ability of tubule cells to lower urine pH, so urine pH may be low or high. If it is high, one might imagine a false diagnosis of dRTA.
This points out in practical terms how the very essence of dRTA is a loss of tubule acidification out of proportion to loss of glomerular filtration. The very name of the disease is renal tubular acidosis, and so pronounced, to overweight and emphasize the specific loss of a tubule vs. a glomerular function. So when filtration is low, one might think everyone would avoid the diagnosis as essentially moot. The problem is that dRTA can lead to kidney disease from all the crystal deposits, and perhaps the many stones, too. Moreover, along their progress to a final death of function, kidneys may pass through transient phases where loss of acidification exceeds loss of filtration, giving rise to periods of disproportionate acid retention.
The Anion Gap Discloses
Blood has many components, but few with both charge and high abundance. Those few are sodium, potassium, bicarbonate, sulfate, and chloride. If you add up the charges on the positive ones – sodium + potassium – and subtract those of the negative ones – sulfate and chloride, you would get the residual anion gap.
But we almost never measure serum sulfate. Instead we calculate the – much larger – anion gap that excludes sulfate from the calculation leaving it – so to speak – as part of the gap.
In dRTA, most of the time, the loss of acid excretion far exceeds the loss of glomerular filtration, so the anion gap – mainly sulfate – remains near normal while the serum bicarbonate – gauged by serum CO2 falls. Essentially chloride ion replaces the lost bicarbonate.
A Way To Picture It
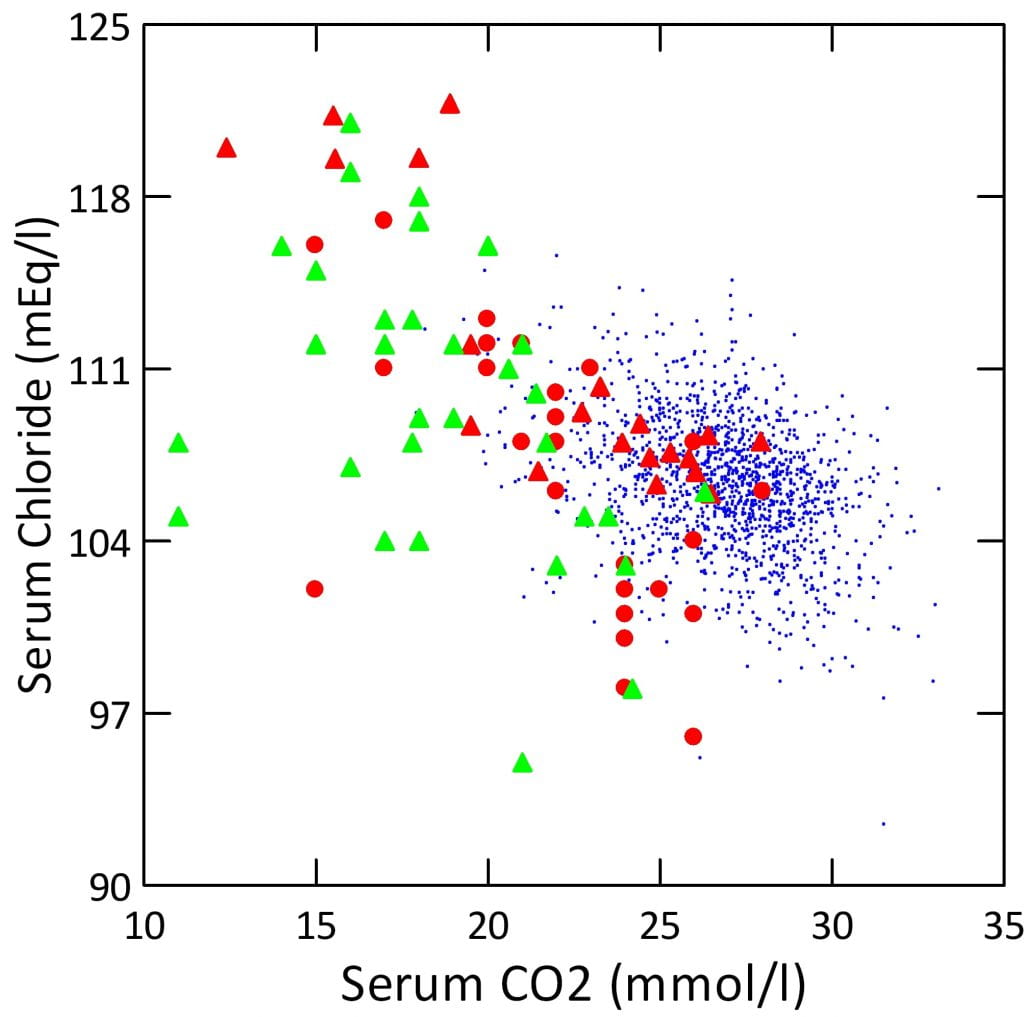
What all this means is that as serum CO2 falls in dRTA, serum chloride should more or less rise in proportion. But when glomerular filtration falls, chloride will not rise as briskly because kidneys no longer excrete sulfate efficiently.
On this plot, tiny blue points are CaOx stone formers. I have omitted other groups for visual clarity. Red triangles and circles are dRTA with creatinine clearances above 60 ml/minute, reasonable filtration. Green triangles are patients with clearances below 60 ml/minute.
For the most part, serum chloride does rise smoothly with fall in serum CO2 when clearance is above 60, and most of the failures – lower serum chloride for a given serum CO2, are from patients with clearance below 60 (green). Serum sodium must play an obvious role, and in fact accounts for the few scattered red points to the lower left.
Clinical Practice
After all this, this labyrinthine odyssey, this thicket of numbers and graphs, what are patients to understand, and what am I telling physicians who are not themselves as particularly interested in dRTA as I am?
Detection and Diagnosis
This disease is detected from fasting serum CO2 and chloride. The former is below normal, the latter runs high so the anion gap is not much above 12 or so. If glomerular filtration is reduced, below 60 ml/min, for example, the gap may have increased.
Diagnosis is the combination of reduced serum CO2 with a urine pH that is not maximally reduced – below 5.3. But in fact, most dRTA produce a urine far higher in pH, usually above 6. The graph I presented earlier of all dRTA cases shows how few have urine pH values below 6 even during acid loading.
Low serum potassium otherwise unexplained – diuretics, laxatives, vomiting – adds weight to the diagnosis. So does a near absence of urine citrate in the 24 hour urine.
Family history of RTA is so obvious a clue I hesitate to mention it here.
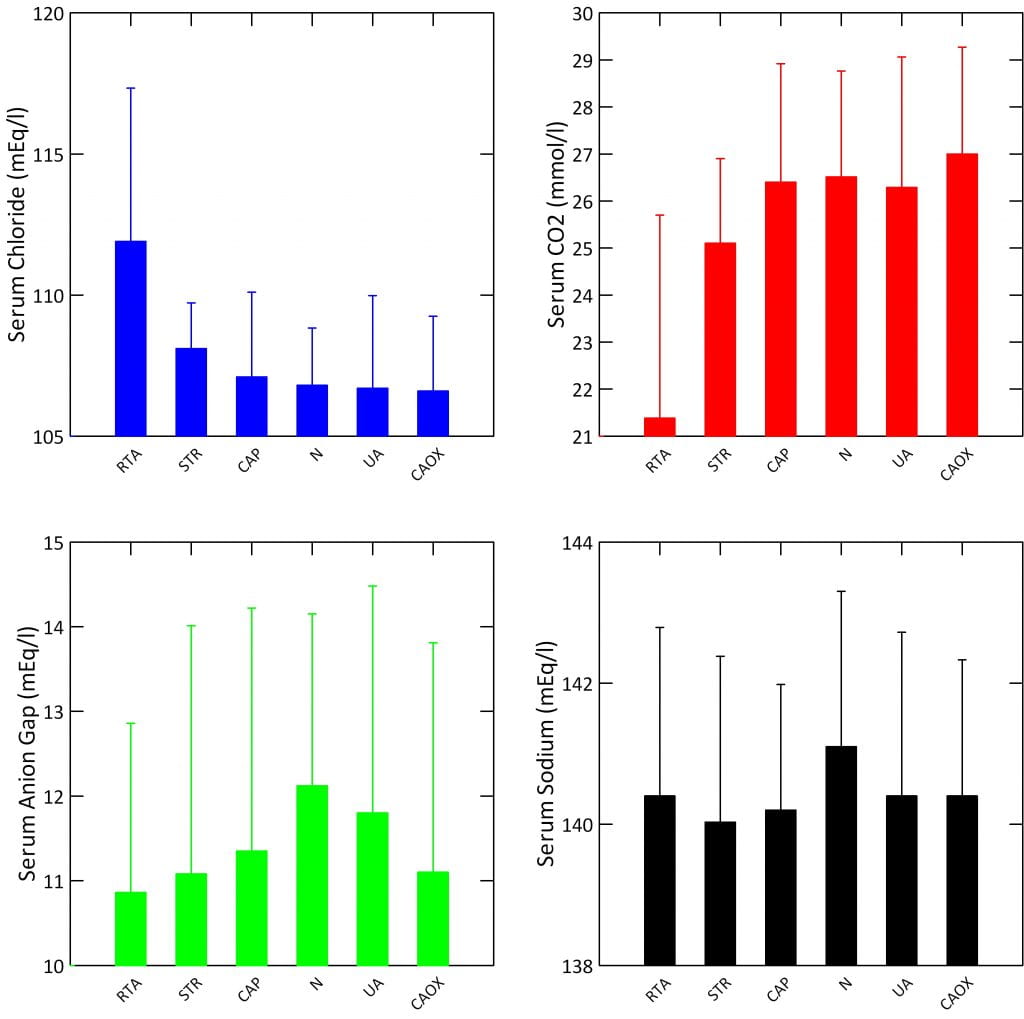
Expected Values for Key Measurements
In fairness, reliable values for serum chloride, CO2, anion gap, and sodium – the last part of the gap calculation – are not readily available for stone forming populations. So I calculated them from my own files, and show mean values here for dRTA, struvite, CaP, uric acid (UA) and CaOx stone formers, along with normals (N). Values are with standard deviations, not standard errors, to show the variability of the values within the populations.
RTA patients are the only ones with low average serum CO2 values, although struvite patients – now that I look at them – are also a bit low. Anion gaps all range between 11 and 12 on average. Serum sodium varies little, between 140 and 141 as a mean.
Since the anion gap and serum sodium in dRTA are like that of all the other conditions, yet the serum CO2 is low, serum chloride must be high – and is.
So it is the balance between serum chloride and CO2 that really swings in dRTA compared to – I guess I can make this sweeping a statement here – all other groups.
Sources of Confusion
If serum CO2 is clearly low – below 23 and preferably even lower, the anion gap is below 12, and 24 hour urine pH is clearly high – above 6, serum potassium is low, and urine citrate low as well, one need not scruple much about the diagnosis except for a few exceptions.
One such is infection with bacteria that hydrolyse urea to ammonia and thereby produce ammonia. Mostly that kind of infection is obvious because urine pH is very high, often above 8, and urine ammonia as well. Another confusion, rather esoteric, is over-ventilation that has reduced CO2 gas pressure and therefore lowered total serum CO2, but raised blood pH. This occurs with normal pregnancy, and can be an artefact during blood drawing – anxiety stimulating hyperventilation. If one confidently excludes these confounders, the combine of low serum CO2 and urine pH above 5.3 – usually higher! – will usually do.
Of course, chronic kidney disease will lower serum CO2, but everyone measures eGFR and will know. Typically, CKD below stage 3b (3a and better) rarely lowers serum CO2 below 23, and when it does urine pH is usually acid and the anion gap may be high. Moreover, contemporary treatment of CKD is to treat low serum CO2 with alkali, so if RTA is present or not one will do the same thing. I have already remarked that along the path to end stage kidney disease acidification may fail to keep up with filtration rate so transient non gap acidosis occurs, and will not explore that topic further.
Definitive Diagnosis
Sometimes some physicians will want to know for sure. One alternative is to administer an oral acid load, typically ammonium chloride, and determine the minimum urine pH achieved. Another is to administer a sodium retaining steroid and a dose of furosemide and determine the lowest pH attained. This reference discusses both. When a young man I did these things, and sometimes published my results, but stopped years ago because almost no patient posed a real problem. Persistently low serum CO2 with an alkaline urine, normal anion gap and low urine citrate is rather obvious. Just the low serum CO2 itself is, after all, something needing remedy. So, I no longer put my patients to the bother of such testing.
Incomplete dRTA
My Own Prejudices
Here is a bafflement for me. Normal blood findings, but if challenged with an acid load these patients cannot acidify their urine as normal people do. Urine citrate may be low, but simple CaP stone formers have lower urine citrate than CaOx stone formers, and I doubt all of them have acidification problems. Excellent scientists have found such cases, and I show points them on my graph as red symbols – meaning failure to lower urine pH fully – with normal serum values. My question is simply what to do about such patients as I nor any other physician can find them sans a provocative test that challenges urine acidification.
I do nothing but treat them as idiopathic calcium stone formers. If low urine citrate is raising stone risk, I attempt to correct it. Likewise for all other stone risks for which we have some confidence: urine volume, calcium, and oxalate.
Recent Science
Burden of Heterozygosity. The brilliant work of Orson Moe showing some people heterozygous for dRTA have impaired acidification tells us how significant the research yield from acid loading tests might be if we all did them. In other words, inability to lower urine pH maximally may point to carrier states for hereditary dRTA. But this offers no specific therapy beyond what we always offer and therefore one cannot justify such testing as patient care but only as research.
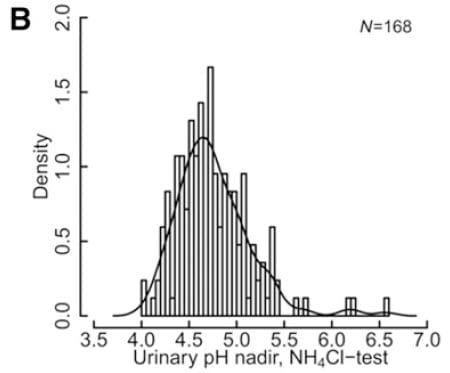 Prospective Study of Minimum Urine pH. A recent and important prospective study of maximal acidification using both acid loading and a more technical method of lowering urine pH suggests incomplete dRTA may be a diagnosis without meaning. This snip from figure 2 of the paper shows one of their main points. The minimum urine pH is a distribution, and 5.3 merely one signpost along the way. In fact the central position of the pH distribution lies at about pH 4.7. No break in the distribution marks pH 5.3 as special.
Prospective Study of Minimum Urine pH. A recent and important prospective study of maximal acidification using both acid loading and a more technical method of lowering urine pH suggests incomplete dRTA may be a diagnosis without meaning. This snip from figure 2 of the paper shows one of their main points. The minimum urine pH is a distribution, and 5.3 merely one signpost along the way. In fact the central position of the pH distribution lies at about pH 4.7. No break in the distribution marks pH 5.3 as special.
Although urine citrate is indeed lower in those with a minimum urine pH below 5.3, a glance at their graph (not reproduced here) of urine citrate excretion and minimum urine pH shows – to my eye – no significant correlation.
Urine calcium excretion is unrelated to minimum urine pH.
The View from the Trenches
I wrote to Dr. Daniel Fuster who has guided this work and he offered his personal view:
‘I think only longitudinal studies that reveal that this subset of patients is indeed unique as it deserves special treatment can rescue provocative loading / idRTA unmasking. Elevated urinary pH, hypocitraturia and low bone mass are also not specific to idRTA but common findings in stone formers and we adapt our treatment to the individual phenotype of the stone former. There is much to be learned about stone formers with high urinary pH. But I am afraid, idRTA as distinct entity in stone formers is likely gone.’
Requiescat in pace.
Causes of dRTA
Being uncommon, dRTA patients invariably attract considerable clinical attention, and the cause of the RTA rarely escapes detection. Given this, I make only primitive remarks about the subject. Textbook chapters and reviews carry long lists of possibilities not appropriate for this kind of overview. A brief and recent one appeals to me for its restraint and focus. But a brief search on PubMed will yield many others as well.
Immune Diseases
Autoimmune diseases (sjogren’s syndrome, lupus, rheumatoid arthritis, and both primary biliary cirrhosis and autoimmune hepatitis) are not rare causes. Renal sarcoidosis and amyloidosis are causes. I have found most of my acquired dRTA among those with Sjogren’s syndrome and recommend this excellent review.
Genetic Causes
In a search for a good review of genetic causes of dRTA, this somewhat older paper deserves attention. Likewise, the remarkable Moe paper on heterozygotes has what seems a complete listing of genetic papers. Family history of dRTA is common in patients with dRTA and a common clue to diagnosis. Conversely, a diagnosis of dRTA should alert families that may harbor more people who have the condition but may not know.
Drugs
Amphotericin B, lithium, and trimethoprim are commonly used and can cause dRTA. Drugs like Topiramate and other carbonic anhydrase inhibitors interfere with acid secretion in the proximal tubule. They produce normal anion gap acidosis, and urine pH can be high, citrate low just as in dRTA. So evaluation must be very sensitive to all medication use.
Treatment of dRTA
So massive an exposition and so narrow a range of treatments!
If any systemic disease is the cause of dRTA, sjogren’s syndrome for example, one treats it as a primary aim. Oftimes, the dRTA can improve, but may not. If a drug is culprit we stop it.
Lacking a systemic disease or drug, which usually means the trait is genetic, or in the event that treatment of systemic disease leaves residual systemic acidosis, one treats with potassium alkali in a dose sufficient to restore serum CO2 to within the normal range. We use potassium, not sodium alkali to restore systemic potassium deficits and avoid raising urine calcium by sodium loading.
If urine calcium is high and does not fall to with treatment of acidosis, I lower diet sodium and, sometimes, urine thiazide diuretics as in idiopathic calcium stone formers. High urine oxalate can be treated, as always, with reduced oxalate intake and increased diet calcium. In other words they are treated as every idiopathic calcium stone former with the exception of a requirement for alkali.
Treatment of dRTA in children is perhaps more urgent than in adults. This form of dRTA is almost always genetic. A single center retrospective analysis of treatment for all patients at a referring center in India. Failure to grow and rickets, were both the rule. With treatment, growth resumed and rickets resolved, in most cases without added vitamin D treatment. In a multivariate analysis only initial growth deficit correlated with eventual height gain – greater deficit, greater gain. Nephrocalcinosis was common and associated only with higher urine calcium excretion. They found that acidosis, not malnutrition was seemingly the main factor affecting growth.
In another article – this one is already too long and tiring – I will review more of the scanty long term outcome data for this disease. But it will add little. We must always correct acidosis, and then use fluids, diet, and our few other means to control stone risk.
A Final Word
Almost no stone formers have dRTA, it is very uncommon. Those told they have it should inquire as to the criteria employed and – I hesitate here – harbor a sceptical sensibility best soothed by a second opinion. Incomplete dRTA seems ridiculous to me and Dr Fuster. It is a name without clinical value in that treatment proceeds as if it were not present. More than ridiculous, it distracts physicians and patients, and can imply to a common calcium phosphate stone former much worse pathology than they have. With him, I recommend we stop using the term outside of research protocols.
Thanks to Joan Parks
The large and colorful graphs of data from our practice all come from a table that Joan made years ago and left me as a legacy.
One might think it a small thing to compile its thousands of rows of data, given we have computers. But that is naive and foolish. One can accumulate considerable volumes of trash from almost any collecting source – like our electronic medical records – but well curated data sets from patient care sources take a long time to clear of error.
In her long career, Joan did this many times.
Although she retired some years ago and now writes excellent novels, we benefit, still, from her legacy of remarkable and accurate research work.
An excellent review, Dr. Coe! And Happy New Year!
Question: Given the reduced citrate:calcium ratio in dRTA (and idiopathic calcium phosphate) and, thus, the need for citrate therapy, do you treat with enough citrate to improve the ratio with no worry about the likely increase in urine pH or is there a particular urine pH at which you feel you can no longer provide citrate therapy?
Hi Stephen, Firstly, glad you like it and Happy New Year to you. In dRTA I almost never get the citrate very high and yes indeed the urine pH goes up – both with potassium citrate. I use it to reverse the metabolic acidosis – required. For residual stone risk after blood total co2 is normal, I try low sodium diet and thiazide to lower urine calcium. That often works. So I get the ratio better by working on the calcium, and I give the citrate because I have to and take what increase I can get as my bonus. There are few of these patients, incidentally, so none of us have a lot of clinical experience. All the best, Fred
Thanks, Dr. Coe.
I guess my question is, assuming urine calcium-reducing therapy is optimized, do you attempt to push the urine citrate level until you reach a particular urine pH (the latter due to concern of the calcium phosphate supersaturation)?
Thanks again.
Hi Steve, Sorry I was not clear enough. Indeed, I do just that. I start with about 30 – 40 mEq/day. Urine pH will go up except in the uric acid stone formers – who usually need more – and citrate may or may not. I judge by SS CaP when I have done what I can. There is a lot of judgment in all this, too, as you well know. For example, if I think urine potassium is low and there is reason to suspect potassium depletion I will use KCl for a while to see if urine citrate rises. Warm Regards, Fred
Hi Dr. Fredric! Thank you so much for the information. What is the normal range of serum CO2 in children? My 3 year old daughter and 1 year old son were diagnosed with dRTA. They take citric acid and sodium citrate 3 times daily. They are gaining weight and growing better. However, I would like to have a second opinion about their diagnosis. Any recommendations in this field?
Thank you so much for your help!!
Hi Dewi, The normal values are as in adults, between 24 and 28 mmol/l; if your children have RTA they need care at a university where the physicians are studying the disease. I do not know where you live but if you say I would be happy to try to find a place for them. Treating the acidosis usually does improve growth and is very important. Regards, Fred Coe
I have the same question. I know someone with dRTA from genetic cause and once we alkalized the urine they developed a calcium phosphate stone.
They are also one a huge dose of potassium citrate and the citrate is still low in the urine.
What is your target urine pH in a patient like this?
Hi David, dRTA is hard to treat. The urine pH is high and usually one can raise urine citrate with a pH change that is itself acceptable. For example the Pka for the second phosphate proton is about 6.8, so if the urine is 7.4, common enough in dRTA, the availability of divalent phosphate will rise only modestly with a pH increase to 7.6 or 7, whereas more citrate can offset calcium phosphate crystal growth and aggregation. It is a hard choice and very dependent on the exact relationship between SS CaP and urine citrate. Best, Fred Coe
Dr. Coe, Your research and writing are brilliant and a godsend. I have Sjogrens, 5 stones. My labs indicate high brushite and oxalate. I will be referring to this article next week with my visit post CT. It’s possible I have dRTA? Would lithotripsty damage my kidneys? Thanking you for you hard work and dedication. Sincerely..
Hi Johanna, Thanks. Given Sjogrens, if proven, dRTA is rather common. Lithotripsy is almost always futile when many small stones are present, as is common in dRTA. Do you indeed have many, and small? You say 5, but is that the total in both kidneys? Given high calcium oxalate and brushite SS, I suspect high urine pH and calcium –
or perhaps you avoid diet calcium and have high urine oxalate. Regards, Fred Coe
Thanks for the information. My 7 year old daughter was diagnosed a year and a half ago when investigating growth issues. Initial labs came back with high urine ph and low bicarbonate, and then an ultrasound showed nephrocalcinosis. Genetic testing came back positive for a dominant mutation of the SLC4AI gene. It breaks my heart to think of a future of kidney stones and pain for her. Potassium citrate 2x’s daily has fixed her acidosis, and she is growing some and feeling better in general. The lack of ability to treat her non-existent citrate and high urine calcium is so frustrating. It feels like we are just waiting for things to get worse. Any proactive ideas for us? We are limiting sodium and trying to get her to drink lots of water. Thanks for your help in this field!
Hi Amy, Your daughter does indeed have genetic RTA and full restoration of blood bicarbonate to normal is important for growth. Lots of water is a good thing. The role of low sodium has never been evaluated but should be either neutral or of benefit. Here is a standard OMIM article on the dominant form of this disease. The main acid transporter AE1 is abnormal in this mutation, and treatment with alkali will be needed long term. The low citrate may be due to potassium deficits, so be sure the dosing is sufficient. Treatment and management of dRTA usually is best done in university kidney stone centers, as the disease is so rare practitioners have little experience. Regards, Fred Coe
I am 32 diagnosised with dRTA with chronic kidney stones. My Maternal grandmother, my mother, and uncle, younger cousin all have this diagnosis. Luckily I believe we are being properly treated after many doctors. I have 2 young children the only testing done so far was a renal panel that came back normal. Do you have any suggestions on what testing can be done for young children? I feel if a diagnosis is given earlier our outcomes will be better.
Hi Sarah, Gene testing for RTA is rather well established, and given the family has so many people in multiple generations I suspect it is an autosomal dominant form. Here is a reasonable review of the genes. Ask your physicians if they can arrange genetic testing of the children. Regards, Fred Coe
Can we do kidney transplant in patients with rta having fair serum creatinine levels but there rickets is making them unmanageable. I saw z patient recently who has his K and acidosis well under control he is 17 years old but because of bone deformities and muscle weakness and retarded growth he is not able to ambulance.
Hello Dr Mahmud, I understand you want to transplant into a patient with RTA whose bone deformities and weakness make it very difficult. I am afraid I would have no answer as to how to proceed practically if the patient cannot be transported by ambulance to the hospital. The rickets is unfortunate as it could have been fixed at the beginning, but now is rather late. I gather renal function has declined so transplant is needed, however, so I can only imagine I would somehow have the family get the patient to it somehow. Regards, Fred Coe
Dear Dr. Coe,
Thank you so much for the writing. My daughter is 2 years old and she was diagnosed dRTA from 1 month old. Now she keeps using bicarbonate & kali every 6 hours and it works rather well on her. I wonder is there anyway this disease can be fixed properly so she can grow up normally as other children?
And now I’m pregnant the second time, is there any test so that I can detect this disease before my child is born?
Thank you again and hope to hear from you.
Hi Mai Le, I answered your email that is a duplicate of this comment and recommended Dr Orson Moe at UT SW medical school. I know he has written back to you and he is an authority on this condition. Regards, Fred Coe
Hi Mai Le, I answered your email that is a duplicate of this comment and recommended Dr Orson Moe at UT SW medical school. I know he has written back to you and he is an authority on this condition. Regards, Fred Coe
thank you my sister have dRTA a24yr old now she have loin pain and uti and stone in ureter -and take ciprofloxacin tab and omnic tab -but sill complain pain what i will to do–
Hi nisrin, RTA is a complex problem. Tell me where you live and I will try to identify an expert for her. Regards, Fred Coe
Dear doctor
Iam 43 yrs old male. Since 2 years i have inflammatory dry eyes, moderate dry mouth, dry skin, atypical face pain in trigeminal nerve region. I never had kidney stones but 3 months ago they found 2 stones each in my left and right kidneys. My rhumetologist thinks i might be seronegative sjogrens. Is my kidney stones likely to be due to dRTA ?
kindly suggest what tests should i do to confirm this condition?
Thanks
Hi Dave, It is indeed not unreasonable. I would do a full kidney stone evaluation and see if the serum and 24 hour urine values agree. One expects high pH, low citrate, perhaps low serum potassium or bicarbonate or both. Lets see. Regards, Fred Coe
I’m so glad to have found this article. I’ve had lowish CO2 for years but it was always brushed off. Then I ended up with metabolic acidosis several months ago. (I believe due to taking neem capsules) The ER doctor diagnosed metabolic acidosis but said it was because I’d been eating low carb the previous two days. He told me to eat carbs. I came in with a CO2 of 5 and left with a CO2 of 7 iirc. I came home and looked up metabolic acidosis and self treated with baking soda. My doctor when he saw my labs was horrified I’d been sent home like that with being given nothing other than IV fluids. Since then my CO2 levels are even lower, running 15-18. My urine ph was 8.5 last it was taken. My chloride levels have run high but not super high. My anion gap has been normal to high. And I have lower creatinine. I’ve also had low potassium but mostly it stays up now because I take potassium chloride on my own daily. I’m finally getting into a nephrologist soon. I’m really hoping for a proper diagnosis. Does dRTA look likely or do I need to have something else in mind? I know I should just trust my doctor on some level. But that’s not always worked out so well for me as you can see. I live in the Atlanta area if you can suggest a good doctor.
Hi Renee, high urine pH, low serum CO2, low serum potassium – sounds like dRTA to me. Your physicians need to confirm this and manage it with you. It is often inherited but there are drugs that cause it, too. You need highly skilled physicians, usually found haunting medical schools; Atlanta has a good one. Regards, Fred Coe
Hi Renee, your story sounds exactly like mine. Did you end up with RTA? Did you get better? x
I am a healthy very active 51 yr old and was diagnosed with bilateral nephrocalcinosos caused by incomplete rta. I am being treated with potassium citrate and hctz, along with low sodium diet and 1 gal water per day. I am not sure if my condition is worsening and was wondering if I need to see a nephrologist or get another opinion in that my urologist is not very informative? I am concerned about my electrolytes and bone health.
Hi Mary, I suspect you form calcium phosphate stones and have calcium deposits in your kidneys, but that is mere guesswork given little real information. I would indeed suggest a nephrologist, who will be perhaps more concerned about bone and electrolyte issues. Regards, Fred Coe
I am a 37 year old female and was just diagnosed with Sjogren’s and dRTA (I also have Hashimotos). I have calcium deposits in my kidneys and have had some removed via lithotripsy. I currently take potassium citrate. I have no other symptoms of Sjogren’s and yet they are recommending that I be treated with Prednisone and plaquenil to see if that improves the dRTA. Do you have any other suggestions? I’d rather not take more medication for my entire life if I can help it. I’m in northern NJ near Morristown. Thanks!
Hi Molly, Sjogren is a systemic disease and can affect kidneys in more ways than dRTA. I suspect they fear effects on the kidney tissues and possible reduction in kidney function. For the stones themselves, I would think water and perhaps alkali to raise citrate – if it will go up – would suffice. But they may have other reasons, and are entirely responsible for your care. So while I do not mention drugs for dRTA, they are used in Sjogren’s and I would do as my physicians recommend. Regards, Fred Coe
My daughter was diagnosed with dRTA at 18 months due to her failure to grow. She has been treated with Bicitra since then. Her father’s family was part of the original study to prove that RTA is a genetic disorder at UCSF in the late 60’s. Her urine pH was usually 9. She has had 12 broken bones, and bilateral osteochondritis dissecans, could these bone issues be related to her dRTA? Also her older sister was told she now has RTA (she is 32 and just diagnosed) because she has uric acid stones. However, she has a urine pH that is normal. She was also never tested for O2, but diagnosed based on her family medical history. Since her symptoms seem to be so different (she had normal growth without bicarbonate), and her doctor said her RTA can go into remission it doesn’t sound the same as my daughters. I told her she should get a nephrologist that specializes in RTA to get a more accurate diagnosis. What are your thoughts?
Hi Tomi, I suspect your child does have dRTA, and IZ presume her physicians will be sure of it and treat it so her bones can heal. Uric acid stones cannot be due to dRTA as they require an acid urine, so I suspect she is entirely different altogether. Be sure the stones are indeed uric acid, and that 24 hour urines show low urine pH as need for them, and her treatment is alkali to raise the pH enough to stop the stones. Regards, Fred Coe
Dear Dr. Coe,
Thank you for you excellent review and I enjoyed reading it. I am wondering if I can get a opinion from you. I am waiting for my appointment with a nephrologist later this month but kinda getting a little bit anxious. I am a 29 year old male and recently visited my primary care following muscle shaking and weakness. The test shows my potassium level was 2.8 mmol/L. Later the urinalysis shows my urine pH is 7.0 and from the potassium/creatinine ratio it seems like excessive potassium waste in urine is very likely. In addition, I did not have vomit, diarrhea or diuretics use. My primary doctor thus suspect dRTA and refered me to a nephrologist. He also mentioned I might be having an undiagnosed stone. What I don’t understand however, is my serum CO2 is consistently almost higher than normal (I did three times last week and it’s always 30.0 or 31.0 mmol/L with 31.0 mmol/L being the highest normal value provided by the lab). The anion gap was 11.0 and is now 7.0 after my serum potassium is corrected by potassium chloride. I am kinda wondering if it is possible that what I am having is an ‘incompelete dRTA’.
I am not sure if it is appropriate to ask for you medical opinion here. In any case, I did enjoy reading this review and learned a lot from it. So thank you very much!
Hi Charles, You have low serum potassium, high urine pH, lots of potassium in the urine and dRTA sounds like a good bet, but your serum CO2 is frankly high. This is surely not dRTA but some other tubule transport defect. I could certainly help figure it out, as these are rather distinctive. If you had a high urine calcium one of the forms of Bartter syndrome would be reasonable. Liddle syndrome causes hypertension which you do not mention whereas forms of Bartter lower blood pressure. But of these, Gitelman syndrome seems most likely. It can begin later in life than Bartter, and commonly presents with muscle symptoms from potassium wasting. Although Bartter and Gitelman both arise from sodium wasting by the renal tubules, they involve different transporter defects. Both can lower serum magnesium, which you have not mentioned. Gitelman has a low urine calcium, Bartter a high urine calcium. Your nephrologist will have no difficulty distinguishing among these alternatives, as they are in every textbook and renal medicine training programs inevitably teach these topics at a high level of detail. The outlook is good but treatment very important. I suggest you do nothing more than take the potassium until your nephrologist determines the cause, and I am sure this will be very straightforward. Regards, Fred Coe
Dear Dr. Coe,
Thank you very much for your reply. If I could just follow up a bit based on what you said – My serum CO2 are not fasting measurements, I am not sure if that changes the picture. I did not have a urine calcium test. My blood pressure seems to be a bit high. I noticed a few higher than 130/80 in my past hospital visits and my father was diagnosed with hypertension in his mid-40s. So I bought a device and measured a few times at home last weekend, I seem to get slightly more than 130/80 a lot, but I am not sure if it is partially due to being a little anxious. My primary doctor sort of dismissed the possbility of high blood presure. My serum magnesium is actually high (2.3 mg/dL with 2.4mg/dL as highest normal, this was when my potassium was 2.8 mmol/L). There are a few other conditions I currently have which I am not sure if they could also be relevant. One is I am seeing a hematologist in the past year for immune thrombocytopenic purpura, the initial onset seems to be in my teenager years but it suddently went lower than 50 last year. So far I have not received any treatment and it seems to be stabalized at 60-70. The other thing is I have been having lower urinary tract symptoms mainly very frequent urination (with small volume each time), which a urologist diagnosed as chronic pelvic pain and i had received some physical therapy. The last time I did an US was May last year mainly to check the spleen but the results also suggests both kidneys looks fine and there were no shadowing calculi.
Anyways, your message is very reassuring. Thanks again for your help!
Hi Charles, You should have more complete 24 hour urine studies. The pain may be nothing or may be crystals from high urine calcium – a Bartter variant. Serum Mg is not always low in Gitelman, so that alternative remains. Let me know what your nephrologist thinks. Regards, Fred Coe
Dear Dr. Coe,
I want to briefly update you about my condition. I have finally met my Nephrologist last week. We felt that the most likely explanation seems to be autoimmune diseaseas such as Sjogrens, considering that I also have Immune thrombocytopenic purpura. However the autoantibodies are all negative at this moment. I am on amiloride now and will see him again in a few weeks. So far he has not mentioned the possibility of genetic defect such as Gitelman, presumably because my potassium from a year ago is normal (I have also found a potassium result when I was 17 and it was normal too). Given these information, do you think Gitelman is still a possibility? I did see a very recent case report (https://www.sciencedirect.com/science/article/abs/pii/S1590865819301124?via%3Dihub) where a patient developed hypokelamia after proton pump inhibitor use and later proved to be Gitleman by genetic test. I did use proton pump inhibitor in December and March as part of the therapy to remove Helicobactor pylori (in the hope that this would improve my platelets at the time).
Thank you very much!
Hi Charles, Your situation is complex and at this stage I would be unhelpful because I lack the details. Genetic testing is possible and this disease can present in strange ways as in your case report. But alas the physicians on the scene have to do the work here. People like me can now consult remotely, legally and with proper information, and if your physicians or you want this my university offers it. But I would hope the local team gets it all done for you – most efficient way. Regards, Fred Coe
Dear Dr. Coe,
I would like to again update you on my case. It has been an interesting experience for me and I learned to appreciated a lot of the things you talked about here. To briefly recap, in August it was discovered my serum K was at 2.8 mEq/L following some mild muscle symptoms. I mentioned before that urine test suggests that renal pottassium wasting is very likely. However I never asked my GP or my nephrologist what calculation they did to determine that this was indeed a renal loss. I took this conclusion for granted based on what I read on this uptodate page (https://www.uptodate.com/contents/evaluation-of-the-adult-patient-with-hypokalemia?search=hypokalemia&source=search_result&selectedTitle=3~150&usage_type=default&display_rank=3#H6814773). I had not had 24 hour urine collection, so based on what was written in the spot urine session, I calculated the pottassium/creatine ratio, and it was about 28mEq/g, from which I concluded it was renal loss. I was on pottassium in the begining but was prescribed Amiloride later by my nephrologist, which is more tolerable. And as I mentioned before, my nephrologist was looking for autoimmune disease that may cause the presumed tubular injury. I also had genetic test done and everything is negative. The conclusion was basically that this was a tubular injury with unknown etiology and I might have to take Amiloride for the rest of my life. However, a few months later I came across an oppotunity to meet with another nephrologist, he suggested that I may not have a kidney issue based on my lab result. He calculated the fraction of excretion for me. Turns out when my serum K was 2.8 mEq/L, the FE was 8%, not so bad. Later, when I was taking pottassium supplement 60mEq a day, the FE increased to 20 % (though my serum K was only 3.5 mEq/L at the time, this kinda still confuses me). Following his instruction, I stopped Amiloride for 5 days and finally did a 24 hour urine collection. The result came back today and my serum K is 3.5 mEq/L, this is in fact basically the same level while I was on Amiloride; the 24 hour urine collection showed about 55 mEq K, which he told me amounts to a 9% FE (I have not seen the creatine number myself yet). He suggests this may simply be a poor diet after all. It’s kinda suprising to me that the two nephrologists are having completely different opnions. I am wondering if you have any thoughts on my case. In any case, I learned a lot reading your website and wanted to share my story. Thanks for reading 🙂
Hi Charles, I gather that in your one sample you had 55 meq of potassium and a serum potassium of 3.5 – I presume fasting! I do not quite recognize a poor diet in that the 55 mEq of potassium surely come from your diet. I would suggest that perhaps the best alternative for you is to stop any meds that might affect potassium – diuretics, amiloride, and potassium supplements – and see if the low serum potassium recurs. You do not mention your blood pressure, but originally I had some idea it might be elevated. Get fasting morning bloods for potassium at reasonable intervals – weekly then if not low monthly or so – for a while and see. If your serum goes down, then get another 24 hour urine and compare the potassium in serum to that in urine. Since selective tubule disorders seem unlikely (the low serum potassium did not recur) possibly you have abnormal aldosterone handling. Perhaps with time the low potassium will recur and your physicians can straighten things out. But the big problem I see here is that measurements were made as meds changed, and one cannot get full clarity. Regards, Fred Coe
Thank you for your article. I’m not a doctor, but I was able to understand it better than any other medical article I’ve read so far!
I am a 61 year old woman. I was diagnosed with incomplete dRTA about 6 years ago after parathyroidectomy. Only after my parathyroidism was cleared up did all hell break loose. Started having kidney stones twice a week. Urinalysis showed that I have both calcium oxalate and calcium phosphate stones. Levels between these to vary from urinalysis to urinalysis. I still have high normal calcium levels. My urine pH was super high. All other blood and urine appear normal. I also have osteopenia. My dad and brother have had kidney stones, but didn’t need further medical intervention. I’m currently taking chlorthalidone 25mg (I cant take more because I have low blood pressure already) and potassium chloride 20MEQ twice a day potassium citrate didn’t do much). I eat a low-oxalate diet. All this seems to be working and I have only 2 small stones now. I’ve had to have lithotripsy twice only.
I was wondering out of curiosity if I actually have incomplete dRTA and should I pay more attention to phosphates in my diet (I drink milk to offset the oxalates). I don’t like dairy and am not a big meat eater. Thank you for any suggestions you might have.
Hi Melissa, incomplete dRTA may not exist, as the article points out. Usually it is a name given to people who form calcium phosphate stones, and have both high urine pH and normal serum bicarbonate and potassium levels, as I suspect you do. I presume your serum calcium is now perfectly normal, and you have the commonplace hypercalciuria often seen after cure of PHPT. You do not mention low urine citrate, which is a remarkable feature of dRTA. Are you really forming new stones or passing old ones? If new, are the stones calcium phosphate? Lithotripsy fragments stones, and the fragments pass causing what seems like more stones. Try to sort that out before launching on into other possibilities. Regards, Fred Coe
Dr Coe,
Thank you for the interesting article! I have Distal Renal Tubular Acidosis. I also have Ankylosing Spondylitis. I started getting kidney stones in 2003 at the age of 28. I took potassium citrate for several years. The urologist took me off of it after several years. In 2010, I started having trouble with UTI’s & pain. I had 14 kidney stones! I now have to have surgery every 3 years to remove kidney stones. I get frequent UTIs. I take 30 mg of Potassium Citrate twice daily! I have a 24-hour urine sample every six months. The Potassium Citrate is helping but still not lowering my ph & raising my citrate. I was told to have a very low sodium diet. I eat mostly Paleo for my Ankylosing Spondylitis. I am now in jeopardy of making other kidney stones because of diet. I try to balance Paleo and the diet I was given to prevent the other stones. Is there anything else I can do?
Hi Alicia, RTA is rare in rheumatoid diseases, but reported. Here is a recent case report. Potassium citrate will raise not lower urine pH, and probably will not much raise urine citrate. Often potassium is lost in the urine and you need potassium supplements, but these can be as potassium chloride/citrate, the latter to keep serum bicarbonate normal. But as with all stone diseases, the issue is urine stone risk and you do not post those. The goal is to keep supersaturation with calcium phosphate below 1, which depends on lowering urine calcium – if high, raising volume, etc. Diet change is directly responsive to what is abnormal in the urine. Regards, Fred Coe
Hello doctor. I was diagnosed with RTA at age 18 during a successful pregnancy. After diagnosis, I have lived a generally very healthy life (age 40 now). I currently take Potassium Citrate 10MEQ ER four times daily. Before diagnosis I saw a urologist that treated my bladder infections (at least 6x/year) and kidney stones (hospitalized for those about once every 2 years). He unfortunately never referred me to a Nephrologist during that time. It wasn’t until my pregnancy that my Potassium level dropped dangerously and I was hospitalized. It was the on-call doctor that put the puzzle pieces together and diagnosed me. I am currently not seeing a Nephrologist, and I should start seeing one again, but I have a couple of questions. I have been told that my kidneys are filled with stones, and I do pass stones on a regular basis with very little pain. Should I be concerned with calcium absorption getting into my bones? My doctor had me have a bone scan about 5 years ago and everything looked good at the time. I believe he was concerned because I went so long without a diagnosis. Should I be concerned now that I keep getting older, should I be taking a calcium supplement at my age with RTA?
I wish I lived closer, I live in Nebraska. I’d love to be a patient. Do you have any recommendations of a nephrologist in Nebraska?
Hi Amy, All these are excellent questions I cannot really answer from such a distance. But telemedicine is here, now, and I can provide care, I believe, even for someone in another state. My secretary Banita Williams 773 – 702 – 1475 can help see if that is possible. If I had a local name for you I would be pleased, but alas. Regards, Fred Coe
Dr. Coe, thank you for the wealth of information here. I passed my first kidney stone several months ago, 50% CaP (apatite) and 50% CaOx, at age 30 (female), with a second non-obstructive stone still in my kidney apparent on CT scan. My metabolic panel was a little bit off with serum Potassium at 3.4 mmol/L and serum CO2 at 21 mmol/L (all other values seemingly normal). Upon doing some research, and with having a history of connective tissue disease (undifferentiated), I came across your information about dRTA. I had a 24 hour urinalysis done (about 1.5 months after the metabolic panel, so values are not paired) concluding a pH of 7.142, slightly elevated Ca excretion (247 mg/d; 4.5 mg/kg/d), supersaturation of both CaP and CaOx (2.34 and 8.04, respectively), and below normal phosphate excretion (0.347 g/d). Other excretion values/ratios were close to optimal and/or were on the low end of the reference ranges. Given my autoimmune history and lab results, I initially suspected possible dRTA, however, my citrate excretion levels were good (761 mg/d) and serum chloride was on the low end of the reference range (101.7 mmol/L). Potassium excretion was normal as well (53 mmol/d). Can dRTA be present with normal citrate and serum chloride values? Or is it possible that everything could be due to a vegan diet (no supplements)? I know veg diets lean towards higher urine pH values, but 7.1 seems excessive. Obviously drinking more fluids will decrease future stone risk, but I need to get that pH lowered as well and make sure there’s not something going on that is damaging my kidneys. I have a referral in for a nephrologist, as I also had protein present in a spot urine sample while passing the stone (which the urologist thought was probably not linked to the stone). If not dRTA or diet, what other conditions and/or tests should I be researching? Thank you!
Hi Caylen, Unclear. The low potassium and CO2 are suspicious as is the high urine pH. But the normal citrate would be odd indeed. The vegan diet will cause a high urine pH, but not a low serum potassium or CO2. I wonder if the serum values have been consistent. Were they fasting? The high SS CaOx (and CaOx stone content) with modest urine calcium makes me wonder if urine oxalate is high or volume low. From here I cannot say much more – not enough about you. I would seek expert consultation given you are young and have a complex medical history. Because of COVID, telemedicine is now permitted and many experts are accessible that way. Regards, Fred Coe
Thank you for the response, Dr. Coe. I have had metabolic panels done fairly regularly since 2017. Prior CO2 values have ranged from 22-25 (mean of 23). Prior potassium values have been at the low end of normal (3.6-3.95). Most recent values 3 months prior to the stone were 23 & 3.95. The only values taken when fasting, however, were the values described above. I have bloodwork due in another week or so and will get the draw done first thing in the morning so that they also represent fasting values. I’m hoping that once my referral goes through I can get a serum P measure as well. Urine oxylate was just barely above ideal (26 mg/d), but volume was a little low (1.56, though I could have sworn it was above 1.7 in 24 hours but perhaps there is some back calculation if my collection was slightly over 24 hours, or I may be mistaken). I ate/drank as I did prior to the stone during the collection. I’m now trying to increase fluids and have stopped putting spinach in smoothies & switched to oat milk from almond milk to reduce oxylate a bit. But the SS CaP & urine pH are still a concern for future stones.
Hi Caylen, If your new serum values are fasting and not normal I would advise seeking specialized medical advice. If you tell me where you live I could try to help. Regards, Fred Coe
New serum CO2 value was still at 21, but potassium is back up to normal at 3.95. Serum creatinine was slightly low (likely due to diet) but everything else in the metabolic panel was within reference range. Do you still feel it might warrant further attention with only bicarbonate being off, and not excessively low? I am in southern Oregon, with no local nephrologist, but telemedicine certainly opens up options (perhaps even consultation with you, if you accept new patients & think you might be of assistance). Thanks again for your insight.
Hi Caylen, You are very smart – I am at the end of what I can offer without more detail etc. Telemedicine is available right now because of COVID and probably will stay around. I am a professor at UC and do see patients remotely. My secretary can arrange a visit and also collect all of your information so we will have it available. Her number is 773 702 1475. Her email is bwilliams15@bsd.uchicago.edu Regards, Fred
Perhaps also relevant is relatively low urine ammonium excretion (18 mmol/day). It’s not below the listed reference range, but is below the 20 mmol/day cutoff I’m seeing used in a few studies the define compromised ammonium excretion levels.
Hi Caylen, It is not easy to interpret urine ammonia excretion in that it varies with urine pH and with acid load – urine sulfate. Regards, Fred Coe
Hello Dr. Coe,
Thank you for your article and sincere look into dRTA. I am not a medical professional, but did find it fairly accessible to me.
I was diagnosed dRTA (genetic) as infant at UCSF and was in a clinical study in the 60’s-70’s. Currently managing the disease with Kcitrate, fluids and diet.
I am interested in longitudinal studies of people with dRTA if there are any. Or is that relevant to my situation as I was one of the first children to be diagnosed at UCSF.
Also, I have significant hearing deficit as first detected before the age of 4 which may be related to dRTA, however there are other factors that may have contributed to the loss. I would like to know if there are any studies that show a progression of hearing loss…all I see are studies on children…and I am interested in over the life time due to dRTA and genes that may be involved.
Thank you.
Hi PB, I know of no studies of the sort you are seeking. But you had your care at the best of places, and seem to have done well. The hearing problem may be related to the dRTA, your gene profile can help with this question. Regards, Fred Coe
My daughter is being treated for distal RTA with Bicitra. Her nephrologist says she is at risk for osteoperosis and so he has started her on another medication called HCTZ. Shouldn’t she be on calcium supplements instead? I read online that HCTZ is a diuretic – won’t that make her need even more calcium? Can you recommend a calcium that is liquid? She is not old enough for pills.
Hi Sue, If your daughter really has this rare condition she should be in care of a university based physician group. It is too rare for even the most dedicated physician to know enough about – and I mean nothing but complete respect for her present physicians. I would suggest asking her/him for help in securing consultation as noted, which will much help in treatment decisions. Regards, Fred Coe
Dr,
Son had gross hematuria at 12 years old. ER missed diagnosis as infection culture came back negative. Followed up with Nephrology and ufology. Discovered simple cyst although he passed a stone doctors refused to look at.
After a few more visits to ER had another stone. Calcium oxalate. Called the lab myself and answer machine said press one for dogs two for patients. No joke. Recently had complete urinary retention again miss diagnosis. Five……Five genetic tests including 3 exomes 1 stone and one polycystic kidney. All neg not family history. Current treatment 3 liters a day water and hydrochlorothiazide. Kidneys extremely small 8 and 12 percent for age. Hypercalcemia idiopathic, bone showing z-2.2. Endo feels no spine fracture not an issue. eGFR unknown cystine c 70 and Swartz 100. Several litholinks showing numbers better still elevated calcium. . PTH normal. I’m so lost. My nephrologist is a good man but any help would be more than appreciated. Numerous KUB’s and US showed a simple cyst. Recently went to low rad dose CT W/o contrast hospital stated 5 stones 2 cysts. Followed by an MRI with one stone and the rest were acquired cysts. All genetics came back negative currently idiopathic. Again all genetic tests negative, no family history and I don’t think I can put him through Anastasia, contrast and a 2 hr MRI again when results inconclusive.
Need help. Can’t let my son die.
Greg
Hi Greg, I gather he has high calcium, but is it hypercalciuria – in the urine – or hypercalcemia- in the blood? I suspect it is genetic hypercalciuria, which is polygenic and not diagnosed via DNA sequencing. The small kidney size is not usual with idiopathic hypercalciuria and stones. I wonder if he has Dent’s disease, which can be diagnosed via further testing. On the other hand, genetic hypercalciuria is a cause of reduced bone mineral. Put another way I suggest his physicians might want to request consultation for him at a university stone center.
Can dRTA be present with normal citrate and serum chloride values? Or is it possible that everything could be due to a vegan diet (no supplements)? I know veg diets lean towards higher urine pH values, but 7.1 seems excessive. Obviously drinking more fluids will decrease future stone risk, but I need to get that pH lowered as well and make sure there’s not something going on that is damaging my kidneys. I have a referral in for a nephrologist, as I also had protein present in a spot urine sample while passing the stone (which the urologist thought was probably not linked to the stone). If not dRTA or diet, what other conditions and/or tests should I be researching? Thank you!
Hi pogba, Normal citrate and serum chloride (and total CO2) are not compatible with dRTA. Vegetarian diets can raise urine pH to 7.1. Incidentally, I normally do not permit comments that carry a commercial link, but your medical question seems legitimate so I have allowed it. Regards, Fred Coe
Hello Dr. Coe,
I’ve been following you for years and thank you so much for all of the work that you post. Grateful. (I’m also a mild classic Ehlers Danlos, mast cell patient with possible autoimmune pancreatitis type2-so I’m a bit of a challenge) I passed a kidney stone about 9 years ago, doctor told me not to worry about oxalates and said the diet was too restrictive. Two years later, my other kidney and tubes were full of stones. Soon the little glass chards, as my doctor refers to them after using the microscope, were coming out of my eyelids, and I developed pelvic pain issues. I quit moderate oxalate foods and added dairy back-major help! I also drank a lot of lemonade for a few years, and have not passed any more stones. But I’m so restricted by my allergic condition that I get hungry and the oxalates make their way back, and I’m miserable; I know it’s gone too far when the little cysts grow on my eyelids. Last year I gently lowered my oxalate foods, and ended up in the ER with elevated chloride and high blood pressure, but anion Gap of 6. I also had severe muscle weakness and cramping. In another event, after trying mild keto for 4 days, my kidneys put out so much fluid, about 6 urinations in an hour for most of the day, I ended up in Urgent Care with no answers. A Genetic test showed possible sodium gated channelopathy, but not certain, but my potassium is usually normal at 3.5, but am on 5 MEQ potassium anyhow per doctors orders. The potassium has really helped my life in so many ways, including muscle cramps, I’ve finally been able to gain a few pounds, and more interestingly it’s helped with autoimmune pancreatitis. I was getting ready for a tough procedure, but it’s now on hold since the pancreas is doing so much better with the potassium. The events where I lose muscle weakness with mild myoclonics, can be scary due to not being able to lift my upper limbs, knee-buckling and not being able to form a sentence for about an hour or so. Overdoing it, caffeine, heat, fasting, too many oxalates can trigger it. They thought of periodic paralysis, but one doctor says no. Today, someone mentioned renal tubular acidosis. But I looked at the anion gap that goes with that, and with mine being low only once during crisis, does that rule out RTA since it is usually an elevated anion Gap? But I did have elevated chloride. Occasionally I will have slightly elevated bun/creatinine, but it usually goes back down. Side note, I also have elevated carnitine, but only eat a few ounces of meat and don’t supplement. The lab sent a note to suggest further workup for CPT1A deficiency and noticed in my research today that CPT1A connected is to RTA, but my doctors didn’t seem to be concerned. Have you come across this connection before of the RTA with the CPT1A deficiency? I’m in my 50’s, so maybe that’s unusual at my age to have this? Also, I feel thirsty sometimes but I drink a lot of water.( No sjogrens or diabetes) Also, only low-dose antihistamines and cromolyn sodium for meds. I have become very sensitive to sodium IV’s, they also cause muscle weakness instantly with eratic blood pressure that is normally low. Thank you so much for listening. Sorry if this is overwhelming. It’s complicated for sure.
Hi Tomesa, ED syndrome is associated with disorders of the renal collecting systems and possibly thereby could promote stones, but a PubMed search for Ehlers Danlos AND kidney stones came up with no useful papers – meaning there are none. I found one paper on renal tubular acidosis and ED but very old and with no followup I suspect it was incorrect (there was medullary sponge kidney mentioned and probably that was also wrong and it was just RTA and ED by chance in one person). A search for CPT1A AND renal tubular acidosis yielded no papers. As for the gene product itself, it is central to lipid metabolism not renal acidification. I wish I could be more helpful, but so far have no immediate clues to how that can be accomplished. Regards, Fred Coe
I wanted to add to my other comment sent a few hours ago, that during the episodes of muscle weakness and very sick feeling that sent me to the ER that my creatinine was low, not high and the estimated GFR was high, but is now back to normal. My urine PH was 5.5, anion Gap low @6, and chloride high @108. Does this have anything to do with oxalates or dumping maybe? I had cut back slowly at the time.
Hi Tomesa, It is helpful. A urine pH of 5.5 makes renal tubular acidosis very unlikely. The only form with low pH is with high serum potassium, contrary to your situation. The low serum creatinine means high glomerular filtration which is also far against any form of kidney disease. Oxalate is itself without any systemic problem apart from stones or perhaps when kidney function is very reduced (not your situation at all). Oxalate ‘dumping’ is a myth – serum levels are tiny and normal kidneys remove the material with high efficiency. In urine the only known problem from oxalate is crystals and stones. Regards, Fred Coe
Hello Dr. Coe,
Interestingly, similar to the commenter above, I also have a rare type of EDS called PVNH EDS, or otopalatodigital syndrome. I came here because I have had chronic stones, burning pain, white cloudy urine, historical urine ph always between 6 and 8, confusion and delirium, lethargy, and chills after urination. I am 5’5″ and weigh 95lbs for my entire adulthood (through childhood I was also consistently in the <1% to 5% percentile for weight-height), and I have been unable to gain weight even though my caloric intake averages 2500 calories daily. This has been ongoing and progressive for 15 years, and I am currently 33 years of age.
I can take 1/4th teaspoon of sodium bicarbonate, and my symptoms will resolve immediately and profoundly, but only for about 72 hours before all symptoms mentioned above returns.
While the stones & urine PH are confirmed by various doctors and objective testing, they have not run an acid load test yet (we've been doing various testing for 6 months now, and the symptoms are fully disabling. I am currently on SSDI disability).
My blood CO2 levels are consistently normal, between 23 and 31.
This sounds like idRTA from my research, because the blood CO2 is normal, while the urine PH has never tested below 6 (however this needs confirmation from acid load testing, which you are rallying against!!).
—
As mentioned, my symptoms are completely disabling, matches idRTA, comorbid with genetic disease and the only known working treatment for me so far is an alkalinizing agent.
Should idRTA be ruled out, even though my symptoms are affecting me this way?
Should acid load tests be truly removed from diagnostic procedures for dRTA, given the above information as it could clarify my condition?
Given the above information, do you believe there is another condition than RTA that matches this cluster of symptoms?
It is also noteworthy that multiple urine cultures have been done and came back negative, and electrolyte levels in urine and blood panels are always normal.
Hi Matt, perhaps the electrolyte measurements include citrate?? Can your physicians calculate GI anion from them: Na K Phos Cl, Ca Mg?? Fred
Hi Matt, otopalatodigital syndrome is not associated with renal tubular acidosis as best I can tell as a PubMed search otopalatodigital syndrome AND kidney stones was negative as was the same with renal, tubular or acidosis, or urine pH. There were some renal anatomical abnormality associations. Your urine is indeed alkaline, but you do not mention the citrate. Is it high or low???Likewise, you do not say what the crystals are (the cloudiness is crystals, I take it). You are better with sodium bicarbonate but that would raise urine pH. I suggest your physicians analyze the crystals and measure urine citrate in the 24 hour urine – RTA it is low, alkali loads high. Acid loading has no place until you have some of the basic information. I consider the problem very complex indeed but suggest crystal type and urine citrate will help. Regards, Fred Coe
Thank you so much for your insight and kindness Dr. Coe!
I was also looking for citrate levels on my urine labs before your reply, but it appears it was not included in the tests. I’m going to ask my doctor about this. You’re the best for taking the time to answer questions here on your articles.
Thanks for this – i landed on this discussion as low urinary citrate and low potassium were noticed upon recent stone formations and blood in urine which ive never had in the past. However, having been diagnosed with the secondary causes by a rheumatologist already this appears yet another new associated problem. Im not sure theres value in looking further into root cause as I started supplemental potassium citrate and my numbers are now coming back normal and no longer having crystals form. Plus as a type 1 diabetic on insulin and i know a little bit about the potassium shift into cells with insulin which in my case makes all this metabolic chemistry very complex. But I will say to my great surprise starting on the potassium citrate it has also very much benefited my blood glucose control as well as my a1c it is actually in normal range now. 🙂
Go figure.
Hi Sara, I gather you have distal RTA from a rheumatoid disease and potassium citrate is helping. That is wonderful and not rare. As for potassium and insulin sensitivity, potassium deficiency is a serious impediment and extra potassium expected to improve glucose control. Regards, Fred Coe
Hi Dr. Coe,
My 5 year old daughter has prominent renal pyramids on scans (nephrocalcinosis ), hypercalcuria, and 3 suspected stones in her kidneys. Her genetic testing came negative and bloodwork is normal. However, with her pH always over 6.5, the FF loading test was done for her and her value did not go below 6 on loading. She was then suggested to be suspected incomplete RTA. I read how you suggest these be treated as idiopathic stone formers. She is on 5 ml alkaliser thrice a day. But hypercalcuria still persistent.
Does this condition with early presentation in children progress to ESRD? Would she benefit from thiazide?. We started her on 12.5 mg daily dose of thiazide and urinary calcium came down in range but on first bloodwork after a month, her lipid profile showed borderline high cholestrol, high LDL, borderline high LDH. The thiazide treatment was discontinued. Since then, her hypercalcuria has returned.
Do you suggest we should still continue thiazide or just stick to alkaliser treatment. Her 24 hour calcium clearance is 4.69 mg/kg/day. Her citrate levels are 1.7 on a range of 1.5-6. oxalates are also high on some reports, however she is negative for genetic conditions on whole exome sequencing.
Hi Anna, This is complicated. Thiazide is used in children with what appears to be idiopathic hypercalciuria. Lipid changes are generally not given much weight vs the problems posed by stones and renal crystallization. I would not think alkali supplements would be ideal as she does not have RTA. But have the gene studies been complete – there are many genetic variants that affect kidney acidification. I would wonder if her physicians might not want to continue thiazide. Regards, Fred Coe
Hi. I’m a 37 year old female. My Mom was diagnosed with CKD about 7 years ago which recently became renal failure at the age of 65. My maternal grandfather also has CKD. If I had to guess I’d say we’ve known about it for maybe 20 years. But he was also born without one of his kidneys (my Mom has both)
I’ve been a bit nervous that this could be a genetic thing in our family. Also in just a little over a year and a half’s time my creatinine GFR went from 117 (sept 2020) to 80 (April 2022) which didn’t help my concern.
Recently I ordered some general health testing from selfdecode and it says I’m more likely to have a lower eGFR, more likely to have a higher creatinine level, higher uric acid and higher total protein.
Because of this I decided to change my diet to mostly Whole Foods plant based. I’m also currently avoiding gluten, dairy, added sugar and eggs. I am eating meat 1 time a week and taking vitamins to fill in nutrient gaps.
5.5 weeks into this diet I got my blood tested again for the first time since April 2022. I was off of vitamins for a couple days and I fasted the day of the lab draw. I also made sure to be adequately hydrated. My creatine GFR improved slightly from 80 to 86. Creatine was 0.89, protein 7.6, anion gap 8. I’m not sure exactly which ones to mention. However I noticed a couple suboptimal values like Chloride being 108 and C02 was 22 (The time before that it was 20). My potassium was 3.8, anion gap was 8. If you’re interested in any other labs let me know.
Anyway, I guess I’m wondering if this is Peru normal or if I should pursue my investigation into a possible genetic cause. The recent labs are what brought me to drta, however I don’t have labs for a urine ph or citrate level. I am wanting to catch and prevent possible kidney problems as I really really don’t want to be like my Mom health wise at the age of 65.
Hi Kalena, I gather you have a gene disorder as per your gene testing (?Healthcode?). Your serum and urine data suggest a form of renal tubular acidosis that is genetic and causes stones and reduced kidney function. Your gene testing needs to be expanded by a medical genetics lab so the exact gene disorder can be specified. Likewise you would benefit from 24 hour urine testing which might reveal high pH and low citrate as part of the syndrome. If you have a genetic cause of kidney disease, treatment may forestall or delay further loss of function. Put another way, you are on the right track. Regards, Fred Coe
hi
my nephew having age 14 months with distal rta
is there some proper treatment because his helth and weekness come with passage of time low patasium leavel and sodium bi corbonate
Hi Mujahid, RTA is a very complex condition and your nephew needs to be under the care of an expert in this disease. Regards, Fred Coe
Hi Dr. Coe,
I have had large (1-3cm) stones for 16 years (started when I was 18). I often pass 5-8mm stones without intervention – I think my body has adapted. They always come back as 100% apatite on lab reports. I was also found to have potassium of 2.6 a few years ago, though I’ve taken potassium supplements since so it’s normal now. Recently I had extensive autoimmune testing done due to liver inflammation, and my SSA and SSB antibodies both came back positive. My urine pH seems okay though – around 6.4/6.5 on Litholink studies, my citrate was 209 and my CO2 seems normal at 24-27. After my last surgery I developed hypertension despite being fit and active – my blood pressure now takes 3 different medications to control and can get as high as 220/130 if not medicated. Do you think it’s worth exploring dRTA? I am reluctant to bring it up lest I seem like a hypochondriac, but I also wonder if there’s something underlying that could also affect my children.
Thank you! This is such a wonderful resource, I appreciate you taking the time to write these articles.
Hi Jacinta. Quite possibly you have renal involvement with acquired dRTA and also kidney disease causing increased BP. I suggest your physicians might want to mount a search for such in case you might be missing your chance for better treatment. Regards, Fred Coe
Hello Dr Coe,
As someone who chooses to be fit an healthy and do exercise regular. Do you have any guidelines on RTA patients.
As someone with likely RTA, I notice my PH drops from 6.7 to 6.4 after gym without fail and also when taking blood test my bicarbonate drops to 21 as normally 25.
Could the lactic acid cause acidosis even further. I’m very interested to hear your thoughts as I could see any info on your site regarding this.
Any information would be appreciated.
Hi Jonathan, If you do indeed have RTA then your blood bicarbonate might respond abnormally to exercise. But urine pH would not specially do so as the genetic defects reduce acidification. Possibly you are heterozygous for RTA or have another disorder altogether. Regards, Fred Coe
Hi Dr. Coe… I was recently hospitalized for shortness of breath with these ABG values: ph 7.52 hco3 13 pco2 18 potassium 3.2 co2 17 Then I was discharged with three days of bicarb and no followup instructions. Then I went home and laid on my stomach for three weeks and regulated my breathing (only through my nose) could not walk or move or talk or breathe hardly at all. I went to another hospital 3 weeks later with SOB still and my ABG values had improved to pco2 26 co2 27 hco3 19 ph 7.48 potassium had dropped 3.2 again…. I was discharged and went home again no medication or follow up.. Actually the only answer I was given was “hyperventilation and anxiety”.
I am now in with a pulmonologist and nephrologist who are both confused by my high blood PH. Have you ever seen RTA in someone with a high blood pH?
I was okay for a month and then all of the sudden acidosis came back right before my menstrual cycle ( I had also eaten processed foods). I went to hospital again and my ABG values were pco2 21 co2 17 hco3 17 ph 7.52 potassium 3.6. Everything had dropped again.. I am wondering is this RTA? My neph says maybe an unusual form of RTA type II. she hasn’t seen high blood ph with this before.
Urine PH over the past few months has been 8, 7 and 6 lowest it has been is 6.
My questions: What is causing my loss of bicarb? is this RTA? Have you seen RTA with high blood ph? Or is this primarily respiratory with a secondary acidosis compensation? My GFR and creatine are normal. I did recently test positive for protein in urine first time recently. I have had ketones in urine also. My pulm function tests, ct scan and vq scan came back as normal.
Hi Johanna, lets look at your blood gas measurements: I have no dates so they are in order
# ph HCO3 pCO2 K
1 7.52 13 18 3.2
2 7.48 19 26 3.2
3 7.52 17 17 3.6
These data are most compatible with primary respiratory alkalosis in which CO2 is being removed at an abnormal rate with the resulting decomposition of bicarbonate into CO2 and increase of pH of blood as bicarbonate goes to CO2 gas as carbonic acid. The blood potassium falls because potassium enters cells, This seems to be acute because with time pH begins to climb back to normal. It has nothing to do with renal tubular acidosis. Regards, Fred Coe
Hello Dr. Coe. I hope this comment finds you well. I’ve found these articles very informative. I am a former nurse (30 years w/ coronary intensive care as my area of specialty) and think I have dRTA Type 1. I am going to be engaging my primary (I’ve had huge potassium wasting in the last several months necessitating really large amounts of supplementation, so I checked my genetics as well which do indicate the susceptibility), and I was hoping you might be able to recommend a nephrologist near central Kentucky, we are fairly equidistant between Louisville, Lexington KY, and Nashville TN. I also have Familial Mediterranean Fever and hypokalemic periodic paralysis.
Any assistance you could lend would be greatly appreciated.
Thanks in advance!
Hi ML, Nicole Miller is at Vanderbilt and a brilliant stone surgeon who is also expert in prevention. She trained in the INDY/Chicago program and I know her well. REgards, Fred Coe
Thank you, Dr. Coe.
First, I want to say thank you for all of the very informative articles you have provided. I just finished the chapter on RTA. Unfortunately, I have had three kidney stone in a period of six months. Prior to that I experienced a stone about 12 years ago (no episodes in between). I was a critical care dietitian for 17 years; thankfully school and career prepared for the biochemistry discussions. My diet for years has been lower sodium than average given the American diet options, lower protein intake per preference, and a daily intake of adequate calcium through milk/yogurt. Stats: 65 year old female, do not smoke and perhaps four ETOH drinks consumed per year, BMI 25.4, bilateral LE congenital lymphedema, slightly hypotensive as norm, A1C 5.
Initially I was informed that stones “just happen” by a primary care provider. I live in a small town without an urologist or nephrologist, but started care with a urology group a month ago (such long wait times to see specialists). I truly expected to be diagnosed with abnormally high oxalate excretion rate and thus a CaOx stone former. Or idiopathic CaPh stone former as that would allow a less restrictive diet going forward. I was not able to catch a stone for analysis and considering my plant based diet intake (higher oxalate intake than average), it made the best statistical educated guess while waiting for definitive testing.
I received my blood work and litholink (24 urine test) results two days ago. Essentially everything on the serum report was normal except a CL of 108 (lab reference high normal was 106); Co2 was 24. (working in critical care ABG was used daily and obviously drawn from an artery, while the result I have was calculated using venous blood). Of note my PTH is on the lower end of normal (19) but Ca is 9 and Phos is 3.6.
For about five years I have had frequent (in excess) urination, increased thirst, drink more (about 3 L water/day), chronic constipation despite fluid intake, daily exercise, lots of fiber. The nighttime frequency increased exponentially this past two years. I completed a fluid study for the primary MD..it quantitated a three day average of 4.5 L fluid intake (food and beverages) and just under 4.5 L output (average void 225 ml). This was not enough fluid for feces, respiration, insensible losses…obviously contributing to constant thirst and constipation.
But! The 24H urine results: Citrate 305, pH 6.682, Mag 40 (normal but low for kidney stone formers) Ca 167 (normal but has a role in RTA) and SSCaP 0.79.
I have extensively reviewed any of the contributing causes of acquired RTA and find that ONLY medication applies to my situation. I take ONE medication: topiramate. I have taken this medication for about 18 years (100 mg every AM).
My follow-up with the urologist is not for another 6 weeks. Here are my questions:
1. Seems obvious to stop the topiramate ASAP (obviously getting a new medication in place first). Remain positive that stopping the medication will resolve /improve the hypocitraturia. Repeat the 24 urine test to assess the urine pH and hypocitraturia post medication change. Or do i wait for the urologist to suggest this…question 2.
2. With topiramate the most likely culprit (GFR 78 and I want to preserve that), a VENOUS Co2 level of 24…..is obtaining an ABG of any significant value in this analysis to delay the stopping of the topiramate? It would mean continuing the medication another 8 weeks, getting an order for an ABG (getting a lab appointment), then starting a new medication in about 9 weeks.
3. I understand the role of Kcitrate in treating hypocitraturia; start taking it? As far as reducing ash (alkali) in the diet my urea nitrogen was 5.34 and PCR was 0.7; I already consume a much lower protein intake than most.
Thank you (truly)
Kristin
Hi Kristin, Topiramate is so powerful a stone forming agent I blame it out of hand. It inhibits carbonic anhydrase with terrible risk for calcium phosphate stones. Please stop it and have your physicians find an alternative. I would do nothing more for at least a month after stopping, then get a new fasting AM blood and 24 hour urine. ABG has no role here unless you have a low CO2 off he med. Regards, Fred Coe
Greetings Dr. Coe,
Thank you for sharing all your expertise. This article is largely beyond me but Jill Harris suggested I post this question here after reviewing my recent 24 hr Litholink results. My citrate fell from the low-mid 600s in 2023 and 2024 to <36 last Ph changed from 6.895 to 5.844 in the same period. I took 4 tablets daily of TheraLith Xr at the advice of my urologist from January 2024 to January 2025 when I discontinued it to see if it was really indicated–I thought not–to see what the results of this most recent LithoLink test. We are stumped, and I have resumed the TheraLith supplements (15 mg B6, 180 mg Magnesium citrate, 180 mg magnesium oxide, and 198 mg potassium citrate / day) due to the alarming drop in citrate.
Other values out of reference range June 2023 – April 2025):
Ca24 370 – 101
Oxalate 36 – 23
Citrate 631- <36
SSCaP 3.18 – 0.32
pH 6.895 – 5.844
Na24 130 – 53
UUN 9.45 – 13.26
Serum results (September 2023 – April 2025)
CO2 22 -21 (avg 17.6 during the previous 5 years 2018 – 2022)
Calcium 9.4 – 10.2
Potassium 4.7 – 3.9
egfr 89 – 53
Would you be so kind as to comment on what next steps you might advise beyond resuming citrate supplementation, and what form and dosage of citrate is advisable once my supply of TheraLith is exhausted in a few weeks.
All my best to you,
Ann
Hi Ann, I gather your urine citrate fell from 600 to unmeasurable over a year or so. A common reason is medication – have you begun any bedications? Bone medications can be a cause of this. Changes during the interval between the normal and the low citrate are most significant to consider. Theralith is just one of many OTC agents to rause urine pH and citrate. Look at everything between the 600 and the low citrate as the cause is there. Fred