 If the lives of the Tudor nobility were luxurious, they were dangerous in equal proportions, for the King who bestowed their riches could in a moment wipe them, and those who possessed them, out. So intimacy with the person of the King and with the whole Royal Family was prized and feared. They lived, these powerful and dangerous people, in their Royal Palaces, to which you must go or have no influence. Even worse, King and Consorts made Royal Progresses, staying here and there as guests of the high nobility. Imagine that, the King as your houseguest. A person, like any other, and yet not at all like any other: glamorous, dangerous, and involved with high concerns.
If the lives of the Tudor nobility were luxurious, they were dangerous in equal proportions, for the King who bestowed their riches could in a moment wipe them, and those who possessed them, out. So intimacy with the person of the King and with the whole Royal Family was prized and feared. They lived, these powerful and dangerous people, in their Royal Palaces, to which you must go or have no influence. Even worse, King and Consorts made Royal Progresses, staying here and there as guests of the high nobility. Imagine that, the King as your houseguest. A person, like any other, and yet not at all like any other: glamorous, dangerous, and involved with high concerns.
You could say this is a silly preface to my common discourse on citrate, but not so. I have written before about its powers in our little domain: It binds calcium, it inhibits crystals, giving it reduces stones. But I have not said how it gets into the urine.
It comes as a royal visitor to some Duke or Marquess, Earl, Viscount, or Baron.
For this molecule has high purposes. It is noble and powerful. What it does in urine is but a tiny fraction of its many actions and probably not one of the more important ones. But what we do when we take citrate calls into play a vast biology. For all our lives we eat a diet that imposes an acid load on our kidneys, our bones, and elsewhere. Our kidneys, especially, adapt to that acid load, so what we call our ‘normal’ state is actually at one extreme. The pills, being alkali, reverse this lifelong adaptation and thereby profoundly alter the physiology of the kidneys and bone. In general one might say the alterations are for the better.
This is a long article but one worth reading for those who prescribe or take potassium citrate pills.
I want to acknowledge the expert error checking of Dr Yangming Cao (UCSF – Fresno) in the section ‘Why are Potassium Citrate Pills an Alkali Load?’ He corrected a significant error in the original article.
A Picture of the Kidney
Many of you are physicians or scientists who know about the kidney, but a few reminders are always worthwhile. Others are neither and we need to have names in common. Human 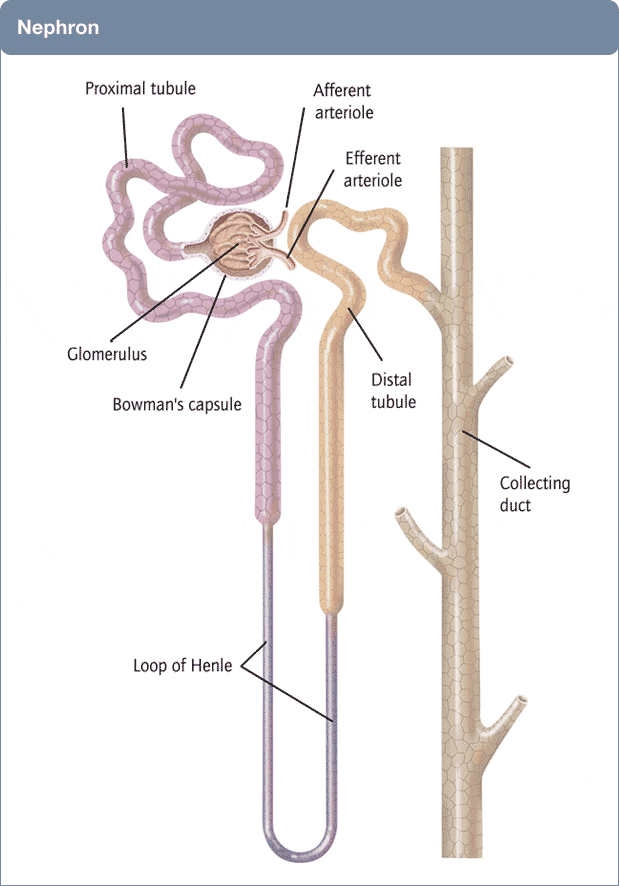 kidneys are made of about one million individual nephron units. The renal process begins with filtration of small molecules like citrate, or atoms like sodium and calcium, through the glomerulus, which is a complex of capillaries whose filtration pressure arises from the heart not a foot away.
kidneys are made of about one million individual nephron units. The renal process begins with filtration of small molecules like citrate, or atoms like sodium and calcium, through the glomerulus, which is a complex of capillaries whose filtration pressure arises from the heart not a foot away.
A majority of the filtered water, salts, and molecules is reabsorbed in the proximal tubule. The distal tubule (highly simplified here) performs tightly regulated absorption or secretion, so as to produce a final urine and maintain blood concentrations in their normal ranges.
These loops will come up again and again on this site so I should comment on the thin and thick portions. The long thin loops of Henle (Henle was the scientist who is credited with describing this part of the kidney) extract water specially well.The thick portions just below the ‘Distal tubule’ notation are called, appropriately enough, the Thick Ascending Limbs of the Loop of Henle. The thick limbs reabsorb NaCl, but not water, and in doing that entrain a marvelous system for – of all things – retaining water! In an article so long as this one, and concerned with citrate, I cannot pause longer here. But we will be back, someday.
Citrate is in the Blood
Kidneys Filter and Reabsorb Citrate
In one published study, concentration of citrate in blood is about 80 – 170 micromolar. A recent review places it at 120 micromoles/liter. If we use 120 micromoles/liter as a reasonable average, and a common value for glomerular filtration of 120 milliliters/minute, the filtration of citrate is about 21 millimoles a day. Of this about 1 – 4 millimoles appear in the urine, the rest being reabsorbed by the kidney cells. So the fraction of filtered citrate excreted is about 5 to 20%, and regulation of this fraction controls the amount of citrate in the urine.
Citrate in Blood Binds Calcium
The concentration in blood of calcium not bound with proteins is about 1 millimole/liter. Citrate concentration is about 0.12 mmol/liter, so in principle about 10% of non -protein bound – calcium can be bound by citrate. Because in calcium citrate crystals 2 citrate molecules can bind 3 calcium atoms, the the figure would seem to rise to to 15%. But in solutions like blood, other materials compete with calcium for a place on citrate – magnesium is one example. So the actual fraction is difficult to estimate. Normally blood citrate level is stable, so although significant, citrate binding of calcium is not likely to influence calcium metabolism by, for example, altering regulation of parathyroid hormone secretion.
Citrate has Signalling Roles
My purposes here are humble purposes, so all I wish to do is put here a tiny list of known effects of citrate on systems throughout the body without pursuing the details. Citrate concentration regulates lipid metabolism via malonyl-CoA. Citrate is sensed by the hypothalamus and thereby affects glucose intake and glucose metabolism by liver. To do these things citrate must enter the relevant cells, and it can do this only via a transporter that takes it across cell membranes.
The Citrate Transporters
NaDC1 and NaDC3
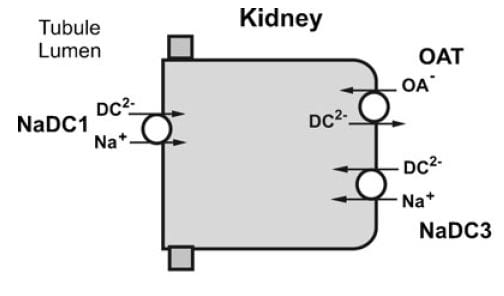
From Pfleugers Arch 466:119, 2014
NaDC1 is on the apical membranes of the proximal tubule cells of the kidney – the surface facing into the tubule fluid – and regulates the rate of reabsorption of the citrate that has been filtered. Its gene is named SLC13A2. This same transporter is on the food side of the small intestine cells and permits absorption of citrate from foods. The featured image for this article shows the structure of the transporter.
The citrate that enters the renal cells can be used for metabolism, or transported out the other side – called the basolateral side, facing the blood – via another transporter called the Organic Acid Transporter (OAT). Yet another transporter, NaDC3, permits citrate to enter kidney cells from blood. Because it appears to regulate urine citrate, my focus is on NaDC1.
The citrate transporter DC1 couples sodium and citrate movement. Since not everyone who reads this will know, let me mention an almost universal property of living cells: they pump sodium out of themselves and pump potassium in. Because they do this, sodium will tend to move into cells if given an opportunity – a hole. DC1 and DC3 can be thought of as sophisticated holes, or channels, through which sodium atoms can move if they have a citrate molecules with them. The actual proportions are 3 sodium atoms move with one citrate molecule, and the form of citrate which moves is one we have encountered before. Recall how citrate binds calcium because each molecule can have 2 or three negative charges on it. The doubly negative (divalent anionic) form of citrate is the one that traverse the channel.
They Transport More than Citrate
NaDC1 permits not only citrate to cross cell membranes but also succinate, alpha ketoglutarate, fumarate, malate, and a variety of less biologically relevant molecules. One might ask why, and I presume it is because the named molecules are all part of the citric acid cycle, which is the main engine of cell energy production. NaDC3 transports all of the same molecules as NaDC1, along with glutarate and a very long list of other molecules not in the citric acid cycle.
This cycle is at the center of that metabolism which uses oxygen to produce energy from food. The reference is to an excellent textbook review that is free online. Another chapter in that book finishes the story of how the cycle produces energy. The antiquity and centrality of the citric acid cycle will become apparent to you if you even browse these chapters. If you read them, you will encounter some of the most important aspects of living cells.
Why are Potassium Citrate Pills an Alkali Load?
In the citric acid cycle citrate is metabolized as citric acid, meaning that 3 protons are taken up from blood with each molecule. Removing protons is identical to adding alkali. Typical dosing is about 20 – 40 mEq of potassium salt daily, but the amount can vary widely.
Commercial potassium citrate contains 1080 mg of the compound in a 10 mEq pill. Typically the potassium citrate salts have a potassium on each of the three anion sites on the citrate molecule. The MW of citrate anion is 189.1. Urocit K, a common commercial version, is a crystalline monohydrate salt so it has a MW of 3×39 (for 3 potassium ions) + 189.1 (for citrate) + 18 for the one water molecule, or 324.1 in all. Given 324.1 for 3 mEq of base, the 10 mEq tablet contains 10/3 x 324.1 or 1080 mg.
The Flow of Citrate
In an earlier era organ physiology was popular and scientists often gathered together 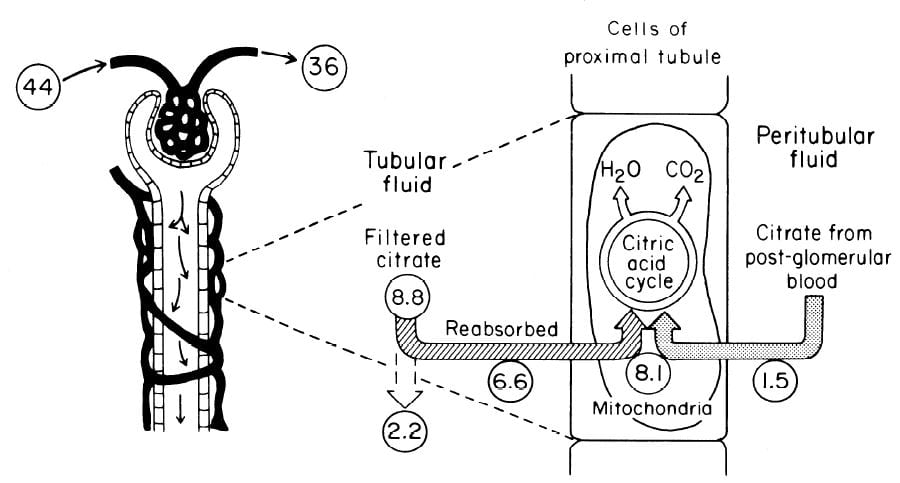 measurements to paint a picture of how things work overall. Here is such a picture from a wonderful review of renal citrate handling by Simpson. Values in small circles are micromoles (umol) per minute.
measurements to paint a picture of how things work overall. Here is such a picture from a wonderful review of renal citrate handling by Simpson. Values in small circles are micromoles (umol) per minute.
Citrate is presented to the glomerular filter at 44 umol/min, and 36 umol/min leaves the glomerulus (8.8 umol/min filtered) in blood what will pass by the blood side of the proximal tubules. From that 36 umol/min, 1.5 umol.min are taken up by renal proximal tubule cells and metabolized in the citric acid cycle. Of the 8.8 umol/min filtered, 6.6 umol/min are taken up on the urine side of the same cells making 8.1 umol/minute for metabolism. The remaining 2 umol/minute (3.17 mmol/day) are lost in the urine. NaDC1 and NaDC3 had not been cloned and sequenced at this early time, but physiologists knew the transporters were there and toted up what they did.
Urine Citrate Varies With Acid Base Status
Acid loads, such as high protein diets, will increase citrate uptake into the renal cells and thereby reduce urine citrate. Alkali loads such as diets high in fruits and vegetables or potassium alkali supplements reduce uptake and increase urine citrate.
Alkali
Clinical Response
In a trial, calcium stone formers with low urine citrate excretion eating a constant diet were given sodium bicarbonate or  potassium citrate, 20 mEq three times a day. Urine citrate rose with both treatments, as did the urine pH. Not relevant here, but in later articles, the sodium alkali did not change urine calcium, but the potassium alkali lowered urine calcium. Alkali itself lowers urine calcium, sodium raises it, and their antagonism is the reason for the differences.
potassium citrate, 20 mEq three times a day. Urine citrate rose with both treatments, as did the urine pH. Not relevant here, but in later articles, the sodium alkali did not change urine calcium, but the potassium alkali lowered urine calcium. Alkali itself lowers urine calcium, sodium raises it, and their antagonism is the reason for the differences.
Mechanism May be Increase of pH
If the citrate transporter is placed into test cells, the movement of citrate can be studied, and such a study shows how powerful is the effect of pH.
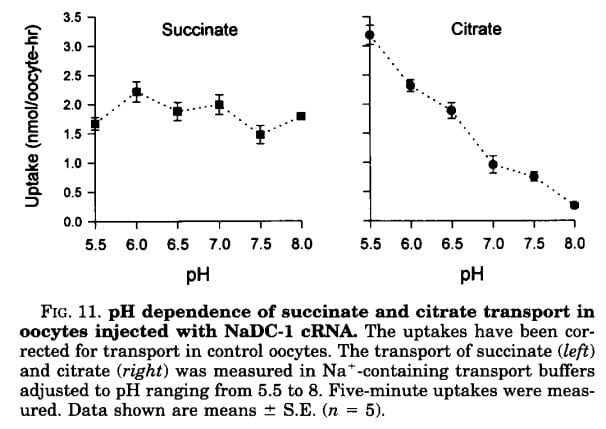 Succinate is a citric acid cycle intermediate like citrate, but its uptake by the citrate transporter is not affected by the acidity or alkalinity of the medium (pH). Citrate uptake is powerfully affected.
Succinate is a citric acid cycle intermediate like citrate, but its uptake by the citrate transporter is not affected by the acidity or alkalinity of the medium (pH). Citrate uptake is powerfully affected.
We have encountered pH before and remind ourselves here that urine values vary from about 4.5 to just below 8. Likewise, citrate has three sites that can accept protons, the acid component of water systems. As I mentioned in the paragraphs just above this point, the charge on the citrate molecule rises with pH as protons are progressively removed, and the sequence of pH values (the pKa values for the dissociating sites for those of you who know about such matters) are 3.13, 4.76, and 6.40. Obviously, in urine, the divalent (2 open negative sites) form will predominate until urine pH rises above 6 and will fall to about 1/2 of the total at 6.4. At about 6.4 transport of citrate was indeed just about half of that at the lowest pH.
pH in the Proximal Tubule
But it is not urine pH which affects citrate transport, it is the pH of filtrate in the proximal tubule of the kidneys, and that pH is not the same as that of the urine. At the end of the proximal tubule, the pH is about 6.7 to 6.8, and at that pH more than half of citrate is in the trivalent form and not available for transport. With alkali loads, as in the experiment in the table, the pH will rise, and citrate transport fall below normal, so citrate appears in the urine.
Problems with the pH Idea
Strangely, modern sources do not mention an older literature which raises questions about this mechanism. Simpson, in an important review from late antiquity (1983), mentions that the drug acetazolamide, which raises pH inside the proximal tubule and lowers pH inside the renal cells raises urine citrate only slightly and at first, but shortly after administration urine citrate falls despite a continuously alkaline urine and presumably tubule fluid. This suggests that even a high tubule fluid pH is not enough to counter the effects of changes in pH within cells or perhaps the blood. So it is not only tubule fluid pH that matters, but perhaps the pH inside the renal proximal tubule cell.
Acid Loads
Those unfamiliar with the matter may not realize that the diet we eat in the US and most of the other first world countries imposes an acid load that must be excreted daily in the urine. So the urine citrate excretion we find in our clinics and in experiments on ‘normal’ diets are those consistent with an acid load. When we give potassium citrate or other alkali we often do little more than neutralize this acid load, yet urine citrate usually rises. Experiments about acid loads add to the diet acid an extra amount of acid.
Tubule Fluid pH
As for alkali loads, a lower proximal tubule fluid pH will increase the fraction of filtered citrate in the divalent form which is transported by NaDC1. The pH of the tubule fluid will fall with acid loads for several reasons. Acid loads – for example a high protein meal – are buffered on blood bicarbonate which lowers the concentration of bicarbonate, and therefore the pH of the filtrate. LIkewise, the tubule cells are stimulated to increase their reabsorption of filtered bicarbonate which further lowers pH. All of this implies that kidneys sense the acidity or alkalinity of the blood, which they surely do.
Transport Adaptation
Over time – many hours to days – the NaDC1 transporter and its gene (SLC13A2) increase 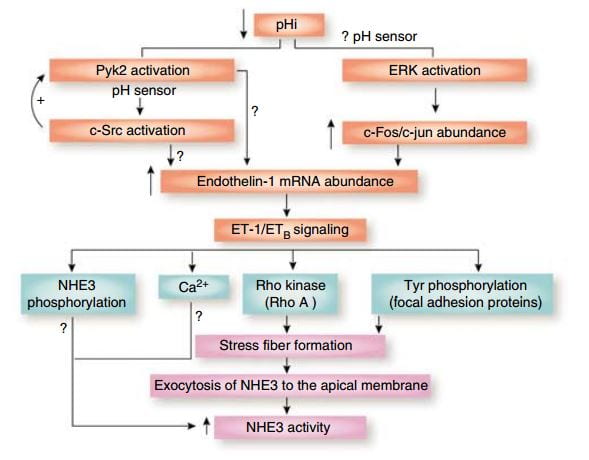 their abundances. This increase is mediated by endothelin – 1 (ET-1) through the endothelin B receptor (ETb).
their abundances. This increase is mediated by endothelin – 1 (ET-1) through the endothelin B receptor (ETb).
This figure from the above reference shows thinking about acid and endothelin as it was in 2007 and seems to be still. A fall in pH in proximal tubule cells can be sensed by a protein named Pyk2, which activates by adding a phosphate to one of its amino acids (tyrosine) and, interacting with another protein (c-Src), increases the abundance of the mRNA of ET – 1 which then signals through its ETb receptor to increase renal acid excretion – bicarbonate reabsorption – via NHE3, a transporter that reabsorbs sodium and secretes acid into the proximal tubule fluid.
This same ET -1 and its ETb receptor also signal increase of NaDC1 transport. Here, mice engineered to have (ETb+/+)or have not (ETb-/-) the receptor were challenged with an acid load. 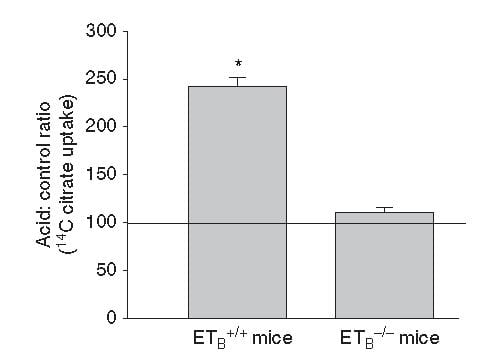 Citrate uptake by isolated NaDC1 transporters in the deficient mice do not respond to acid.
Citrate uptake by isolated NaDC1 transporters in the deficient mice do not respond to acid.
So one and the same effect, acid sensing and endothelin – 1 signalling increases acid excretion and citrate conservation.
But, you may ask, why am I grouping these two together?
It is because both concern acid base balance.
Citrate is metabolized as citric acid, taking up 3 protons per molecule metabolized, which is the same as saying it provides 3 molecules of alkali – like bicarbonate. Loss of citrate is therefore loss of potential alkali. NHE3 is a main driver of acid – protons – out of blood into proximal tubule fluid which reclaims filtered bicarbonate – conserving alkali.
So urine citrate, which we are interested in because it binds calcium and inhibits crystals, has a much larger role to play – part of the grand system which maintains a constant blood pH against the acid or base loads of diet.
Which pH?
I have spoken about pH of the proximal tubule fluid, of the blood, of the urine, but the one that is central to regulation of NaDC1 is the pH inside the proximal tubule cells. That pH appears to respond to acid or alkali loads, but the manner of its response is not simple. The signalling is through the Pyk-2 sensor already discussed and a parallel pathway via ERK (same diagram, above) which I did not discuss. But how sensing works, what is sensed, this remains very much an open research issues, and I will leave off here as this article was about urine citrate and the conversation has already taken us through many byways, beautiful if exhausting to follow.
Potassium
But – that awful word – one important fact remains to be uttered. Depletion of potassium lowers the pH inside kidney cells and lowers urine citrate. I will not pursue the details of this well worn story, except to point out its extreme clinical relevance. Diuretics that are used in stone prevention, or for hypertension, deplete cell potassium stores. It is the potassium citrate we give to patients.
Ammonium, and the Rest of the Story
How can I leave off without filling out the details of how kidney cells respond to acid challenge with production of ammonia that balances acid load with acid excretion?
Bicarbonate
A Better Buffer than Most
A buffer keeps pH relatively constant by taking up protons when they enter a solution and giving them up when alkali enters. It is a kind of shock absorber.
At the beginning, evolution favored bicarbonate. It is a buffer of considerable virtue in that it can take up protons or release them, like common buffers do, but has a special trait.
Bicarbonate is forever in equilibrium with carbon dioxide gas (CO2). When bicarbonate takes on a proton to become carbonic acid, much of that acid becomes carbon dioxide gas. When protons are taken out of blood, CO2 gas forms new carbonic acid which donates a new proton to the solution, and essentially bicarbonate appears in solution ‘out of thin air’. That it flows from solution into thin air and back makes bicarbonate a more stable buffer than those which live only in solution so it was an excellent choice.
What Kidneys do with Bicarbonate
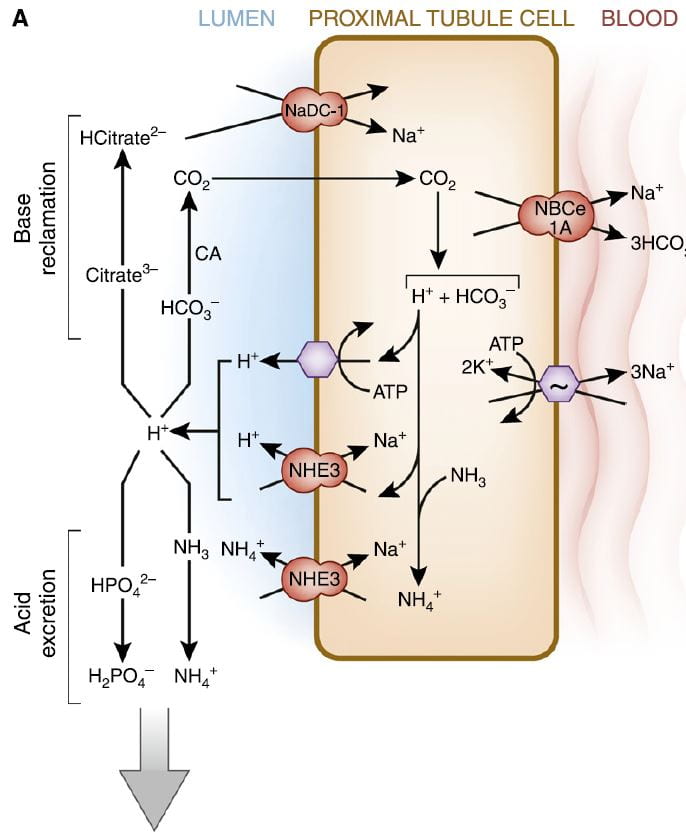 It is this very molecule, bicarbonate, which the kidneys traffic in when they respond to alkali or acid loads, and it is, of course, CO2 the lungs regulate in blood under the control of the brain.
It is this very molecule, bicarbonate, which the kidneys traffic in when they respond to alkali or acid loads, and it is, of course, CO2 the lungs regulate in blood under the control of the brain.
The figure is from the ‘A’ panel of a lovely drawing in a lively and engaging review. Being small, bicarbonate is filtered, and being the main buffer of the blood almost all of what is filtered must be reclaimed. So the proximal tubule cells, which do most of that reclamation, busy themselves forever with that task.
The way they do it is the simplest way. They add protons (H+) to bicarbonate in the tubule fluid, which becomes, as I have said, carbonic acid that transforms into carbon dioxide (CO2), which gas passes through the cell walls into the interior. Note, ‘CA’ is carbonic anhydrase an enzyme which speeds up the process of the transformation. In the cell, the CO2 becomes carbonic acid. Because protons are being pumped into the tubule fluid, protons are stripped off the carbonic acid so it becomes bicarbonate. The bicarbonate enters the blood with Na via the NBCe1A transporter.
There are two proton pumps. One uses ATP for energy to move the protons. The other (NHE3) uses the low Na in the cell as a gradient; sodium moves in through a channel like a revolving door, which makes one proton go out for every Na that moves in. At the blood side of the cell, the ancient ‘Great’ ATPase pumps Na out and potassium in, as it does in most cells that live on Earth. NHE3, the exchanger, is the molecule we met a few paragraphs above. It is increased by Endothelin 1 via the ET1b receptor.
At the top of the left side of the picture is citrate, our little slice of this massive structure. A few scraps of proton add to citrate so it has 2, not 3 negative sites, and can be reabsorbed. Its gene is regulated by endothelin 1 so when NHE3 is increased so is NaDC1.
Phosphate
Reclaiming bicarbonate is Sisyphean work. Nothing happens to get rid of acid loads from meals. But more protons are secreted than are needed to reclaim bicarbonate. Some are buffered on phosphate. But all the protons buffered on phosphate produce bicarbonate from carbonic acid inside the cell, and that bicarbonate enters the blood via NBCe1A.
Ammonium Ion
Ammonia is produced in the proximal tubule by removal of nitrogen from glutamine, pictured at left. As always, 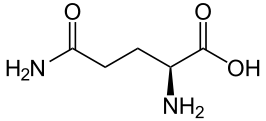 kinks are carbon atoms in this kind of drawing. The first one on the left has an oxygen and NH2 group, and a bond to the next carbon. Carbons typically form 4 bonds each. The next 2 carbons are merely linked to one another. The fourth has another NH2 and the final one at the right 2 oxygens. The left hand one is removed by an enzyme to produce NH3 and glutamic acid. The second one is removed to produce α-Ketogluteric acid which lacks any NH3. The 5 carbon skeleton remains unchanged.
kinks are carbon atoms in this kind of drawing. The first one on the left has an oxygen and NH2 group, and a bond to the next carbon. Carbons typically form 4 bonds each. The next 2 carbons are merely linked to one another. The fourth has another NH2 and the final one at the right 2 oxygens. The left hand one is removed by an enzyme to produce NH3 and glutamic acid. The second one is removed to produce α-Ketogluteric acid which lacks any NH3. The 5 carbon skeleton remains unchanged.
Ammonia (NH3) can tale up a proton to form NH4+, ammonium ion, which has a pKa of 9.3 meaning that at the pH of proximal tubules and cells, it is fully protonated. Loss of this ammonium ion in urine represents net acid excretion because the protons that were taken up came from carbonic acid which is converted to bicarbonate and transported into blood. Unlike titration of phosphate, excretion of ammonium ion does not increase urine pH because the pK is far above the pH of urine.
Under normal meal conditions, about 40 – 60 mmol/day of acid are excreted, of which about 2/3 is ammonium. Large acid loads, as for example, a ketogenic diet for weight loss, would induce a large increase in ammonia production so acid excretion can keep pace with acid production.
α-Ketogluterate
One might think this byproduct of glutamine metabolism, the 5 carbon skeleton, might be metabolized and done with, but no. A significant amount is metabolized. But some is not.
What is not metabolized traverses the kidney to cells in the later nephron, the intercalated cells in the collecting ducts, which usually pump protons into the tubule fluid to create the final urine p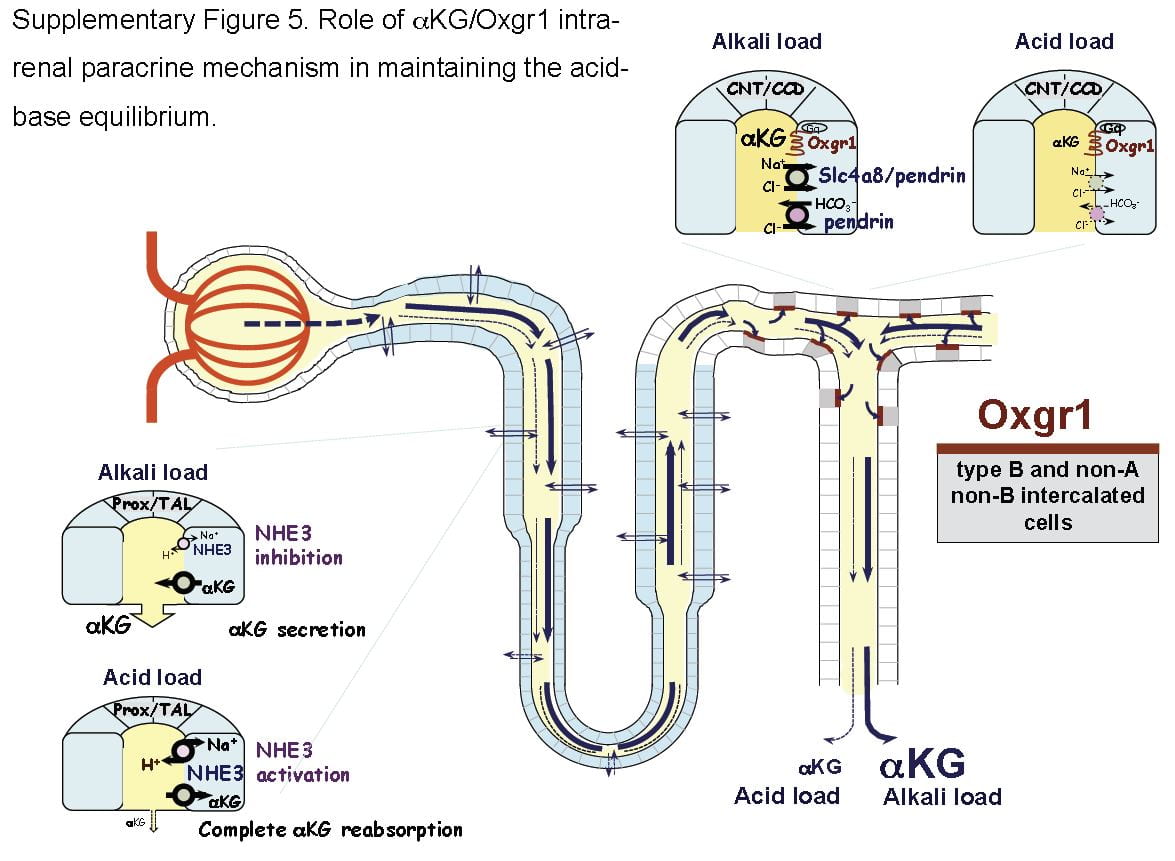 H which is critical to supersaturation and stone formation. But these same cells can reverse themselves and pump bicarbonate into the tubule fluid and protons into the blood, and they do this when confronted by an alkali load.
H which is critical to supersaturation and stone formation. But these same cells can reverse themselves and pump bicarbonate into the tubule fluid and protons into the blood, and they do this when confronted by an alkali load.
It turns out that α-Ketogluterate is itself filtered and reabsorbed in proximal tubule, and its reabsorption is profoundly reduced under alkali conditions so that more is delivered distally to a receptor (Oxgr1). When occupied by α-Ketogluterate this receptor signals the reversed intercalated cells (B and non-A cells) to increase their secretion of bicarbonate. The transporter for α-Ketogluterate is NaDC1. The net effect is to enhance bicarbonate – alkali – loss which offsets alkali loads.
The same receptor signalling stimulates pendrin, a complex exchanger which moves bicarbonate and Na together with chloride to effect NaCl and NaHCO3 reabsorption. Because acute acid challenge increases and acute base loading reduces proximal tubule NaCl reabsorption, this action would tend to maintain salt balance in that the intercalated cells would increase salt reabsorption as proximal tubule reduces salt reabsorption. Of note, although chronic acid challenge increases NHE3 abundance and activity, it reduces NaCl reabsorption via effects on other transporters. For these reasons the α-Ketogluterate – pendrin link is probably more important in minute to minute or hour to hour regulation than in adaptation to acid or base loading diets or treatments.
Citrate and Oxalate
You would think I had exhausted the topic by now, but no. NaDC1 and slc26a6, the citrate transporter and the anion 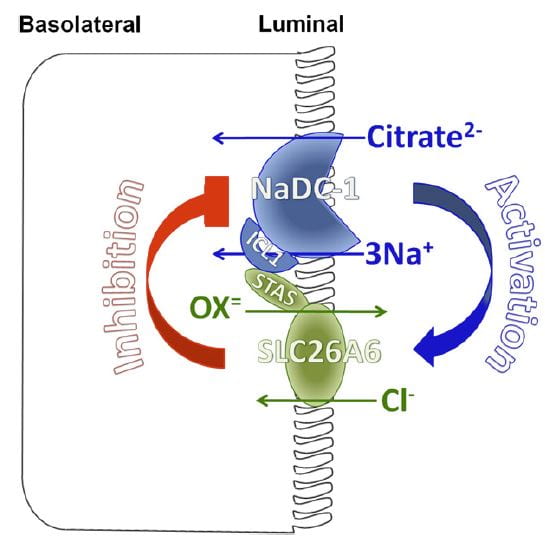 transporter (oxalate is an anion it can transport) which disengages NaCl transport from NHE3, themselves interact in relation to kidney stone formation.
transporter (oxalate is an anion it can transport) which disengages NaCl transport from NHE3, themselves interact in relation to kidney stone formation.
At least in animals and in cell experiments, the two transporters – which are present in a complex within the renal cell membrane – interact as in the figure. Slc26a6 inhibits NaDC1, so that when actively transporting oxalate into tubule fluid citrate reabsorption is reduced, urine citrate rises, and binds urine calcium to reduce risk of calcium oxalate stones. When oxalate secretion is minimal, NaDC1 increases to salvage citrate.
These animal and cell experiments imply that in human urine citrate and oxalate excretions should show parallel changes; this has not been tested.
Putting it All Together
Just Diet
Our urine citrate is an outcome of our biologies, which are variable, and our diets. Most of us eat a diet that imposes a net acid load, so our kidneys tend to conserve citrate and α-Ketogluterate, our intercalated cells pump protons not bicarbonate in to the final urine, our proximal tubules produce considerable ammonia and our urine pH is about 5 – 6.
Some of us, vegetarians whose diets do not have a proper balance of protein, very massive fruit eaters, as examples, have low citrate reabsorptions and high distal deliveries of α-Ketogluterate; our intercalated cells are reversed and stimulated to put bicarbonate into the final urine, our proximal tubules do not make much ammonia.
But the words ‘most’ and ‘some’ are misleading. In the US, certainly, chronic acid loading is the overwhelming rule, and the same throughout Europe and considerable parts of urban Asia. So our ‘normal’ poise centers on adaptations to acid load. It is not that we live in a neutral acid base condition, demanding from our kidneys little excretion of acid or of alkali. Life long we demand acid excretion. That is where we start. It is to that task our kidneys – and our bones, as I shall someday speak about – apply themselves all the days of our lives. However it is, for good or for evil, that lifelong adaptation to acid load affects us, that is our state, our permanent condition.
What Does Normal Mean?
When we give potassium citrate or any other alkali in doses of 40 to 60 mmol/day we neutralize a large fraction of diet acid. This is best considered not so much as an ‘alkali load’ as it is the removal of that acid load to which we have long been adapted.
Of course, urine citrate rises. Because we give alkali over months or even years, renal cells will adapt fully to the changes. But by ‘adapt’ I mean they give up the adaptations to acid loading. In the case where the dose of alkali just matches acid production one might best say the kidneys are relieved of their burdens in either direction, and reveal the way they would function if not driven to either extreme.
Like the small sailor plying the simple waters of a bay fills its sails sometimes southerly, sometimes northerly, making little way, dancing before a playful breeze, the cells shift their powerful machinery a bit here or there as one meal gives way to another. What shall I call this state of freedom? Why is this not the ‘normal’ from which point we register the responses to extra alkali or acid?
I have read where it was in Eden a condition of fruit, as the animals were not for them to eat. Perhaps I am wrong, and if Eden was as it says in our books the ‘normal’ state was alkali load. Perhaps Milton is wrong. After all he was not there, merely a poet making into life what he read in a holy book.
Potassium Citrate Pills
Raise Urine Citrate and pH
The expected changes are a decrease of proximal tubule reabsorption through reversal of the effects of chronic acid load. ET-1 signalling must fall, citrate reabsorption must fall because NaDC1 is no longer stimulated by ET-1 and because proximal tubule fluid pH will rise and with it the fraction of trivalent negative citrate.
Urine bicarbonate and urine pH will also rise. Partly, blood bicarbonate will rise and with it filtrate bicarbonate concentration and pH. NHE3 transport will be decreased vs. chronic diet acid loading, the baseline in the first world countries, and much of the proton secretion will be used in reclamation of bicarbonate. Naturally, NH3 production will be greatly reduced because the Pyk-2 sensing system will be signalling a higher pH.
Increases in Citrate and pH Vary Among People
But the biology is complex enough that in some people the main response will be citrate, and in other bicarbonate. Given all of the regulatory steps and signalling pathways involved a variety of responses is inevitable. Clinically this means one must measure and determine if the main effect is mainly increase of citrate excretion or of pH and therefore of CaP SS.
What is the Ideal Dose?
A nimble answer is enough to match net acid production – urine sulfate excretion is a decent index. I suspect that answer because of the problem of high urine pH in some people, and because as a clinician I never find it perfectly suits most patients. Yet it is a good starting dose because it aims at neutral acid base balance.
A Simple Pill with Powerful Effects
Physicians who treat kidney stones may well be the main ones who prescribe alkali loads to people with normal kidney function over months or even years or decades of life. This is indeed a remarkable physiological and clinical experiment, and that we do it makes the physiology and cell biology of acid base balance a central topic in clinical practice of stone prevention.
Likewise patients who take this humble medicine undergo what amounts to a reversal of cultural norm, which is a condition of chronic acid loading.
Thence, and for this reason, I have written a very long article about the topic, for physicians and their patients, and especially for scientists who know more about this topic than I do but may not see things from exactly the same view point.
G’day Dr. Coe,
I was diagnosed with Sjogren’s disease in 2015 and in 2016 when I had CT of my lung, it was suspected medullary nephrocalcinosis in both kidneys. The recent ultrasound of the abdomen (March 3, 2022) says that there are hyperchoic foci with posterior shawdowing filling both renal collecting system shape – S/o staghorn calculus of both kidneys, less than 1.5 cm sized two anechoic cysts seen in the right kidney, no evidence of hydronephrosis. Test result of Feb, 2022:
The urine test result: ph. 7, S.G. 1.005, Leucocytes +, WBC 9-13 (standard range is 0-3;
CHE: Ureal 50.4mg/dL; Creatinine 1.3mg/dL; Na. 142mmol/L; Potassium 2.76 mmol/L; CL 114.1kU/L;
HEM: WBC3.5, RBC 3.71, Hematocrit 36.7%, PCT 0.146%, Monocyte 12.6%;
SER: Vitamin B12 510.1pg/mL.
My potassium has been below the standard level since 2016 when it has fallen down 1.4. Ever since I take Potassium chloride (KCl) 7.5 solution 30 ml/day. As a result it keeps around 2.6-2.7 with reaching 3.2 last year, but decreased again in Feb. 2022.
My doctor prescribed me a table that makes PH down and I had it for 3 months. but the urine PH is still at 7. PH 7 has been kept for the several years.
At this moment can’t tell what type is my kidney stones. Here 24 hour urine test is not available. ESWL is not available either.
In addition, I have stomach acid reflux.
So my question is what would be your advice to reduce kidney stone. Will potassium citrate work for me? Any other advice would much appreciate. Thank you.
Hi Chimge, You have Sjogren disease diagnosed in 2015 and your potassium was low by a year later and stays down despite potassium chloride. I gather your serum creatinine is 1.3 mg/dl so kidney function is reduced a bit. Urine pH is very high and your serum chloride is high. I suspect you have renal tubular acidosis from your Sjogren disease – a common consequence – that is causing the high urine pH, renal potassium wasting, and kidney stones – which will be calcium phosphate. The disease damages cells that acidify your urine – thence the high pH and potassium wasting. It also reduces kidney function. Agents to acidify the urine cannot work and may worsen things. I would ask your physicians if they wish to use potassium citrate to improve your blood acid base status (the high chloride surely reflects a low serum total CO2 content) and consider treatments for the kidney disease itself. Your condition is not simple, and perhaps they might want to obtain consultation from a university program convenient to you. Regards, Fred Coe
I have hypoaldosteronism, parathyroid hyperplasia (in remission/post-surgery) and possible MSK per latest imaging scans. I have made some stones all my life and they are mostly ca-ox. They got very bad for a year before the parathyroid operation. I just had another bilateral ureteroscopy for stone removal and a second surgery because both kidneys blocked and I am recovering after the stent removals. I take potassium gluconate to manage my depletion from the low ox diet and effects of fludrocortisone. I use a portable ECG and measure my ST curve and BP to adjust. My urologist wants to add potassium citrate but I cannot figure out what impact that will have on my other conditions or how to dose it. In the summer, I take upwards to .5 or .6mg and have serious potassium depletion from that and sweating. I can get away with 1/3 to 1/2 that under lower stress conditions. Should I do magnesium citrate instead? I cannot do a 48-hour collection for another month probably. I have tried potassium citrate a couple of times and do not seem to tolerate it. I feel like I am doing something wrong or that I should be doing something different. Any tips would be appreciated.
Hi MG, I find your situation confusing. I gather your physicians have established that you do not produce sufficient aldosterone and are treating you with a hormone that mimics some of the actions of aldosterone. I also gather you have considerable potassium wasting from the hormone. A low oxalate diet does not lower serum potassium – at least that is something certain. As well you have had PT surgery and have stones perhaps ascribed to hyperparathyroidism. If things are to the point you use your own ECG monitor for management, your physicians may wish to seek expert consultation that might help simplify your life. Certainly one might reconsider the dosing of the steroid so as to reduce potassium wasting. Given the complexities of 24 hour urine interpretation with potassium depletion, your urologist must feel a bit confused. Magnesium citrate has no apparent place in your management. Regards, Fred Coe
Hi Dr Coe
My husband just started taking potassium citrate to prevent and treat his uric acid kidney stones. He has a J-pouch (as a result of having had ulcerative colitis for years with extra intestinal manifestations) and he often takes loperamide to reduce the number of bowel movements.
I would like to know if loperamide would be contra-indicated with potassium citrate.
Thank you in advance
Hi Amal, The drug reduced intestinal motility and the routine pills are large so I have concerns. But more – diarrhea fluids are alkaline and high in sodium, so often the correct alkali is sodium bicarbonate. The 24 hour urine is valuable here in that urine sodium is often quite low making a sodium alkali preferable and – incidentally – completely safe. Perhaps his physicians might want to review his 24 hour urine studies and decide if this is a good thing. If not, perhaps the liquid forms of potassium citrate would be safer than the pills. Regards, Fred Coe
Hello Dr. Coe –
I’m trying to work through the math you provided in this article – about the calculation of mEq from 1080 mg of potassium citrate. As I understand the conversion formula, it is (amount of potassium citrate in mg) / (molecular weight of potassium citrate) X valence of potassium citrate. In this case, that would be 1080 (weight in mg) divided by 324 (mol. weight), times 1 (valence) = 3.33 mEq. – not 10 mEq! Am I doing the calculation correctly? Or I missing an additional factor?
Hi Dr Ratner, The valence of citrate in the crystal is 3, so it is 3.33 x 3 (slight rounding errors get you to the 1080). Best, Fred
Thanks, Dr. Coe. That now makes sense! Understanding the chemistry is challenging…and the concept of valence is tricky. I have an additional question – relating to the efficacy of ‘citric acid’ alone. You’ve written that unless citric acid is complexed to a cation ( i.e. K, Mg, etc.) it will not alkalinize the urine or reduce crystallization. However, isn’t there evidence that certain powdered lemonade products (i.e. Crystal Light) – can effectively reduce calcium oxalate stone formation? And, if that is the case, is that because they contain citric acid?
Hi Dr Ratner, it is because at the pH of that preparation a significant fraction of the citric acid is dissociated – the pKa values range from around 3, 4.7 and 6.4. The anionic from of citric acid, which must exist in solution as an anion salt of corresponding cations like K and Na, will generate bicarbonate as it takes up a proton to metabolize in the cycle that bears its name. Fred
Dr. Coe –
I believe you have said that citric acid ‘alone’ (i.e. not as a salt with Na,K or Mg) will not act as an alkali, and won’t raise urine pH – that it might just get processed in the Krebs cycle.
Does this mean that the enthusiasm for ‘lemonade’ or “crystal light” for stone prevention is misplaced?
Hello Dr Ratner, No. In the lemonade, depending on pH, some of the citric acid will he in the anion form. It has 3 pKa values: 3.13, 4.76, 6.40. So in blood it is fully dissociated, and in urine kidneys can titrate down to the second pKa making it a divalent contributor to titratable acidity. In beverages, pH hovers around 3 (very acid!) to perhaps 4.5 so citric acid is significantly ionized and capable of being a proton receptor. The pH of Crystal Light is such that it contains 20 mEq of citrate ion per liter. For lemons themselves, it will depend on how ripe. pH runs low in lemon juice, about 2, so most of the citric acid is protonated. Dean Assimos and colleagues measured the amounts of citric acid in numerous fruit beverages and found a lot in fresh lemon juice but did not report the pH. Recently, John Asplin has noted much more citrate in orange juice drinks and the low sugar ones are reasonable K cit tablet alternatives. Crystal Light lemonade pH and citrate were measured by John and I put the main facts in my site article. Regards, Fred
Dr Coe, quite a remarkable write-up on potassium citrate—thanks so much.
My urologist wants to put me on potassium citrate, ramping up to 40mEq. I have Celiac Disease and Sjogrens Syndrome, and this amount of potassium citrate concerns me since my GI tract does not absorb well. I have calcium oxalate stones. I had two this year, but in general it is about nine or ten years between stones. They range in size between 2mm and 6mm, most of the time around 3mm.
Wouldn’t 10mEq be enough to help? I am all for preventative, but there is a bigger picture here with complications from my autoimmune diseases.
Pete
Hi Pete, You are not simple. Celiac disease and Sjogren both can cause stones and for different reasons, so the 24 hour urine and serum studies need really careful scrutiny. I presume your urine pH is high and citrate low – from the Sjogren, or citrate low and pH low (from the celiac disease), but I am just guessing. As to dosing, in either case you need alkali enough to raise citrate and also treatment of any other urine abnormalities (oxalate?, calcium? volume?). The dose is what is needed to achieve the goal. Sodium alkali can work, with some effort, and foods and beverages can help get what you need. The type of stone gives clues as well. That is about as far as I can help given what I was told. Regards, Fred Coe
Dear Dr. Coe,
I have been taking potassium citrate tablets as supplements over multiple years (in some countries they are treated like that). The reasons were that I had neural symptoms from low potassium, and low potassium was measured in a hospital.
I have lowered the dosis a lot to now just 60mg of potassium / 100 mg of citrate per day, after I changed my blood pressure medication from ramipril to elanapril, which seems to let me keep my potassium much higher.
Now it seems I have acidosis, my sweat is like acid, my throat is sore and my kidneys and some muscles are hurting, a few hours after intake of normal Asian acidic food. No worries, I will see a doctor tomorrow.
I was reading your article and understood that my kidneys might have been protected from acid through my potassium citrate intake, and they might have gotten used to that protection. I will have to switch to alkali nutrition and hope that my kidneys get used to living without potassium citrate. Is that realistic? How long can that take? How much are my kidneys damaged that their acid reduction capacity is so low? So far the only thing that was found with them is a little scaring and several benign cysts.
Hi Greg, You cannot really assess kidney function subjectively. If you are concerned your physician can measure your kidney function and blood alkalinity and tell you if there is a problem. But what is the disease that caused low potassium?? That is not a common condition. I would think the cause would have a name and specific treatment. Long term alkali use will not alter kidney function appreciably. Regards, Fred Coe
Is it normal for the Potassium Citrate (I’m on 10meq 2 tab 3x a day) to come out in stool completely whole/solid? Am I still absorbing it? Thanks!
Hi Cathy Ann, Yes. The wax matrix stays, the drug is absorbed. Fred
Hello most respected Dr. Fredric Coe.
I just wanted to inform you that my 24 hour urine analysis showed that I had ABOVE reference value for citrate and at the time I was taking zinc and magnesium citrate just by chance for zinc/magnesium supplementation and also because for me magnesium is great to help pass stool and also a physician informed me it can help with migraines. It seems like other things than potassium citrate could be useful for loading up on citrate in the urine?? Magnesium citrate especially is very readily available and also affordable, and its side-effect profile is vert low. In fact I even noted my first red-coloured stone ever when I stopped supplementing with zinc citrate and magnesium for a while, which made me also go back here as red colour apparently indicates it’s more of a uric acid stone. My other stones have been dark and brown mostly calcium oxalate with some phosphate stones!! So the red one was a first and that makes me think citrate and urine pH levels got too low from stopping zinc citrate and magnesium supplementation! I’ve also read on pubmed that there is evidence for magnesium itself helping to counter stone formation, so maybe magnesium citrate could have extra double useful effects from both the magnesium in itself together with the citrate?
Would be nice to hear what you think and if you think MDs should maybe be researching more into this? And I guess not just magnesium itself but the fact that citrate can be loaded in more ways than fruits and potassium citrate supplementation etc, and that strange lemon soft drink powder you have over there in the US.
Hi Anton, Your remarks are very sound but if you produce red stones one would need to analyze them to be sure they are uric acid and not something else. Additives may be fine for stones but only 24 hour urine testing can tell if that is true – they are the basic foundation along with stone analysis. So I think you should get fully tested as in the link and then see if the supplements are the right thing or not. Regards, Fred Coe
How would I find how much elemental citrate is in 1000mg of calcium citrate and how much elemental citrate is in Urocit-K 15meq (1620mg)? Is there a formula for this? Any help would be appreciated. Thank you
Hi John, calcium citrate is 3 calcium atoms with 2 citrate molecules. Calcium AW is 40 mg/mmol, citrate is 192 mg/mmol of the acid. Given this, the weight of 2 mmol of citrate as tricalcium citrate is 2×192 + 3×40 or 384 + 120 = 504 mg. So a 500 mg tablet of calcium citrate provides 2 mmol of citrate. The good news is that citric acid has three proton sites, so the 2 mmol is 6 mEq. Your question: 1000 mg of calcium citrate is 12 mEq of citrate and of course the Urocit K is 15 mEq of citrate. But note that you are getting a LOT of calcium, and that could affect urine calcium and stone risk. Ask your physician about that. Regards, Fred Coe
I take Potassium Citrate in supplement form, 200mg per tab and I take 10-14 tabs daily. It has been a godsend for my interstitial cystitis. I came upon your article while trying to find out if it also alkalizes my stomach acid? Also, should I be concerned taking this level of potassium citrate long term? Thanks!
Hi SLR, Your dose is 2 mEq 10 x a day or 20 mEq a day. That is perfectly safe, and I am glad it helps with cystitis. I was not aware that it did so. Perhaps it reduces crystal formation there. It does not alter stomach acid. Regards, Fred Coe
Hello Dr. Coe,
I have a bladder stone. Is it possible to reduce its size through taking Potassium Citrate?
Hi Paul, if it is uric acid, yes. One can tell from the CT density (your physician can easily measure it). If it is calcium based, I am afraid not. Regards, Fred Coe
Husband is stage 2b CKD with normal potassium blood test. Nephrologist had him increase his potassium citrate 1080mg from 2 to 3 pills a day. Why take potassium is it’s normal and with CKD? He hasn’t had an oxalate kidney stone in years since changing diet.
TIA
Hi Gina, stage 2b requires no special diet or treatment, per se. There is some literature showing a protective effect of alkali in slowing progression of kidney disease. Probably that is why his physician is using this medication. Regards, Fred Coe
I have many kidney stones since 1988 passes at 1.1cm level . lab says that is calcium oxalate stones . will it be helpful taking pottassium citrate -XR tablets
Hi Sisira, Not necessarily. All depends on what is causing your stones. Here is my best take on how to find that out and plan effective prevention. Regards, Fred Coe
Hello Dr. Coe,
I had PCNL surgery less than a year ago. 70% of the stones were calcium phosphate and 30% were calcium oxalate.
The results of two 24 hour urine tests about 5 months apart are the following:
Vol 24 SS CaOx Ca 24 Ox24 Cit24 SS CaP pH SSUA UA24
7//22 2.03 2.51 50 22 215 0.16 5.962 0.43 0.372
12/22 3.39 1.96 104 32 516 0.72 7.321 0.02 0.513
After the 7/22 24 hour urine test, I have been taking 15meq potassium citrate two times per day.
Would you advise that I continue with the 30meq potassium citrate per day?
Hi Alan, I have some trouble aligning your data. I think it is volume 2.03, 3.39; CaOx SS 2.51,1.96; Ca24 50, 104; Ox24 22,32; Cit24 215, 516; pH 5.9,7.3; and SS CaP 0.16,0.72, 1/22, 12/22/22. If so, you began the K citrate between the two collections. Of course, the conditions of these collections cannot be an accurate reflection of those when you formed stones because CaP SS is <1 by some margin, and SS CaOx very low in terms of stone risk. Partly this reflects what seems a very low urine calcium. My suspicion: You lost some kidney function after the PCNL, urine calcium fell, and SS with it. This would show up as a rising serum creatinine from before the surgery. Right now you seem to have no stone risk at all without the K cit or with it, but given stones are CaP perhaps you might be better off without it. Your physicians are in charge here, so do not act on my comments but perhaps you might want to mention them to your physicians. Regards, Fred Coe
Seeking advice– pt on 10 mEq K citrate BID, w mildly high urinary citrate (1.3 g/D) and mildly high urinary ca (333 mg/D). My thought is to leave the alkali dosing unchanged and encourage the pt to reduce dietary sodium. To put this another way, what is the risk to the patient of “excess” urinary citrate levels. THANK YOU
Hi Dr Kleinschmidt, I apologize for the errors that have led to such a late reply. The site has been redone with the attendant confusions. Raising pH with citrate may raise SS with respect to calcium phosphate – as brushite – and provoke more stones. So it is not the citrate but the pH that worries me. Lower diet sodium will lower urine calcium and perhaps lower SS CaP but the strategy seems less than the most simple one. I might just focus on the urine calcium and use only enough alkali to bring urine citrate above 400 mg/d – the threshold at which increased stone risk begin. Regards, Fred
Hi,
Do you recommend the powder or capsule version? And if powdered, how much? I have been reading 1 tsp. or so.
Hi Suzanne, The pills are about 1080 mg each of K citrate giving 10 mEq of alkali. Usually 2 to 4 daily are needed if one is trying to raise urine citrate. The powder (food grade) is potassium citrate crystals and 1000 mg needs to be weighed out with a food balance. A teaspoon is far too much. Because potassium is not entirely without risk your physician must be involved in its use for stone prevention. Regards, Fred Coe
Hi
I have a URINE PH of 5 and have had a very high oxlate diet of the past 2 years. I lost aroiund 33kg on Keto and Carnivore. I am diagnosed with Psoriatric Arthritus and gout, but only take proben 500 mg per day. no other meds. I exercise 7 days a week, around 40 to 60 mins per day. My CREAT A level vary a bit due to exercise and diet, but ,my CYSTATIN levels are at 0.67, and 121 EGFR (normal GFR 67, CREAT 113) as of March 2023. If I hydrate well and reduce PROT, I get my CREAT down to 100 and my GFR up to 78. I am trying to reduce oxalates to protct my kidney and have eliminated nut butters, reduced Whole Whaet bread from 4 slises to 2. Reduced Bulgar Wheat to 200 grams per day. I find that certain veg caise my extreme pain in my lower legs and ankles, I have no idea why. If I stop them, the discomfort goes away so I avoid veg now. I am also worried because probenecid has kidney stones a side effect. Never had a kidney stone and I am 46. I take CALcoum (500mg), VTA D (1000 IU), VITA k (45mcg), POT CITRATE (100 mg) and MAGN Glycinate (100mg). POT and MAG only started recently since I read about POT CITRATE Kidney protecting properties. What advice do you have, do you think I am on track. My TRIG levels are 0.6, Coles normal, Calc normal, Mag normal, Pot normal, Sodium Normal, all electrolytes in fact. Folic acid normal, B12 normal. I have extensive tests which I am happy to share.
Hi Marius, As you do not have stones I am not clear in what way I can be helpful. I can say that potassium citrate is not proven to protect kidneys. It can raise urine pH and prevent uric acid stones, but you have not made such stones. So your issues are about diet in general, and while reasonable and seemingly complex beyond what I can offer on this site. Best, Fred
Dear Dr. Coe, thank you very much for all the information that you share.
My husband had 2 ureteroscopies for calcium oxalate stones before knowing the KDS diet. These were created by a high oxalate and low calcium diet.
Now he has been following the KDS diet pretty well for about 3 weeks.
Today’s 24 urine test shows lower oxalates: from 70 mg/24h to 38, but calcium raised from 368 to 404 mg/24h
The citric acid is high: 1131 mg/24h
The rest of the labs are normal.
The last scanner shows he still have stones with one of 7 mm
He has been taking potassium citrate for abut 5 years. (currently 99mg 2 a day, but before he was at 99 mg a day)
Should he stop taking the potassium? And do you think he is on the right track?
Hi Jacqueline, His urine calcium is very high. Presumably it is the idiopathic hypercalciuria of stone formers.The high citrate can occur with diabetes or pre-diabetes, or may be from his potassium citrate. I presume his blood calcium is normal – very important. Likewise his serum phosphate is not low. I imagine his physicians will attend to these issues, but I should mention them just in case. Regards, Fred Coe
Is potassium and magnesium chloride included as a stone-forming salts?
Hi KG, tje 24 hour urine panel for stones included potassium and magnesium but not because either ion produces calcium based stone crystals. Magnesium combines with oxalate lowering its availability for calcium. Potassium is a major part of urine ionic strength which latter controls the activity coefficients for mono and divalent (charge numbers) ions which one needs to calculate saturation. Clinicians use potassium and magnesium levels to deteect and evaluate a number of systemic causes of stones and to calculate GI anion (alkali) uptake. Regards, Fred Coe
Dr. Coe,
I have had 3 kidney stone surgeries. Lithotripsy 2x which left stone fragments that grew in to more stones and recently ureteroscopy. Hopefully I am “stone free” as my Doctor and I are focusing on prevention now. I am now on potassium citrate after a 48 hour urine collection. I am experiencing GERD symptoms with this medicine and came across a product called stone stopper that also is the same equivalent of potassium citrate but claims to be easier on the gut. Is this a reasonable substitution?
Thank-you
D. Krupinski
Hi Debra, Given the pace of your stones perhaps they are composed of calcium phosphate in which case potassium citrate would be a problematic choice. Did your 24 hour urine show a low citrate? I suspect it did and also an increased calcium and pH. I really do not know anything about your situation so these are just remarks you might use in looking at your own labs and perhaps discussing with your physician. As for stone stopper, I reviewed some of these OTC preparations. Most use sodium instead of potassium to permit OTC distribution. I do not believe your potion was on my list. Regards, Fred Coe
i have more regular episodes of gout as ive aged, more painfull and longer lasting .ive tried allupirinol and colcichine which helps slightly do you think by adding potassium citrate will regularise uric acid levels?
Hi Wahid, I know of no evidence that oral alkali will help with gout. Fred
I had a laser lithotripsy for a calcium oxalate monohydrate kidney stone and after a couple of 24 hour urine tests was told to add alkali citrate to my diet. I subsequently started taking Moonstone. I also have been taking famotidine for acid reflux for many years. I have developed peripheral neuropathy from a vitamin B12 deficiency. I believe the vitamin deficiency may have been promoted by the lack of stomach acid resulting from the famotidine. I was wondering if the Moonstone could also have contributed to a low acid environment in my stomach. I have read conflicting articles on this. One said “
“Alkali citrate supplements work by providing citrate, which binds to minerals in the blood after they are absorbed from the intestines. This can indirectly create a slightly more alkaline environment in the body overall. However, they don’t directly neutralize stomach acid. Overall, while alkali citrate may have a slight alkalizing effect, it is unlikely to significantly alter the highly regulated acidic environment of the stomach.”
While a 2nd report said:
“citrate acts as an alkaline compound in the digestive tract. When ingested, it neutralizes stomach acid, potentially providing relief from acid-related discomfort”
Can you shed any light on whether I need to be concerned about Moonstone/alkali citrate increasing my stomach pH enough that I don’t process vitamin B12 correctly?
Thanks.
Hi Gary, I can indeed. Citrate is an alkali because it is metabolized as citric acid, meaning it takes up three protons from blood buffers as it enter cells as an energy source (in the citric acid cycle) and produces no protons as it is metabolized. The proton loss means new bicarbonate buffer is produced from dissolved blood CO2. Your problems are from the proton blocker. Potassium citrate can take up protons from the stomach but comes in wax delayed action form that prevents such. Potassium citrate crystals themselves could act as a buffer in the stomach. Regards, Fred Coe
Does Moonstone have the wax delayed action? Or do I need to switch to citrate pills?
Hi Gary, Moonstone does not have delayed release than I know of. Fred
Hi Gary, I can indeed. Citrate is an alkali because it is metabolized as citric acid, meaning it takes up three protons from blood buffers as it enter cells as an energy source (in the citric acid cycle) and produces no protons as it is metabolized. The proton loss means new bicarbonate buffer is produced from dissolved blood CO2. Your problems are from the proton blocker. Potassium citrate can take up protons from the stomach but comes in wax delayed action form that prevents such. Potassium citrate crystals themselves could act as a buffer in the stomach. Regards, Fred Coe
Hello Dr. Coe –
All the studies use dosages in mEQ, but supplements are sold in mg. What is the conversion?
Thx,
Dom
Hi Dom, Here is an article on OTC alkali. It has things in weight units. For K citrate 1080 mg is 10 mEq of base. Regards, Fred Coe