 If the lives of the Tudor nobility were luxurious, they were dangerous in equal proportions, for the King who bestowed their riches could in a moment wipe them, and those who possessed them, out. So intimacy with the person of the King and with the whole Royal Family was prized and feared. They lived, these powerful and dangerous people, in their Royal Palaces, to which you must go or have no influence. Even worse, King and Consorts made Royal Progresses, staying here and there as guests of the high nobility. Imagine that, the King as your houseguest. A person, like any other, and yet not at all like any other: glamorous, dangerous, and involved with high concerns.
If the lives of the Tudor nobility were luxurious, they were dangerous in equal proportions, for the King who bestowed their riches could in a moment wipe them, and those who possessed them, out. So intimacy with the person of the King and with the whole Royal Family was prized and feared. They lived, these powerful and dangerous people, in their Royal Palaces, to which you must go or have no influence. Even worse, King and Consorts made Royal Progresses, staying here and there as guests of the high nobility. Imagine that, the King as your houseguest. A person, like any other, and yet not at all like any other: glamorous, dangerous, and involved with high concerns.
You could say this is a silly preface to my common discourse on citrate, but not so. I have written before about its powers in our little domain: It binds calcium, it inhibits crystals, giving it reduces stones. But I have not said how it gets into the urine.
It comes as a royal visitor to some Duke or Marquess, Earl, Viscount, or Baron.
For this molecule has high purposes. It is noble and powerful. What it does in urine is but a tiny fraction of its many actions and probably not one of the more important ones. But what we do when we take citrate calls into play a vast biology. For all our lives we eat a diet that imposes an acid load on our kidneys, our bones, and elsewhere. Our kidneys, especially, adapt to that acid load, so what we call our ‘normal’ state is actually at one extreme. The pills, being alkali, reverse this lifelong adaptation and thereby profoundly alter the physiology of the kidneys and bone. In general one might say the alterations are for the better.
This is a long article but one worth reading for those who prescribe or take potassium citrate pills.
I want to acknowledge the expert error checking of Dr Yangming Cao (UCSF – Fresno) in the section ‘Why are Potassium Citrate Pills an Alkali Load?’ He corrected a significant error in the original article.
A Picture of the Kidney
Many of you are physicians or scientists who know about the kidney, but a few reminders are always worthwhile. Others are neither and we need to have names in common. Human 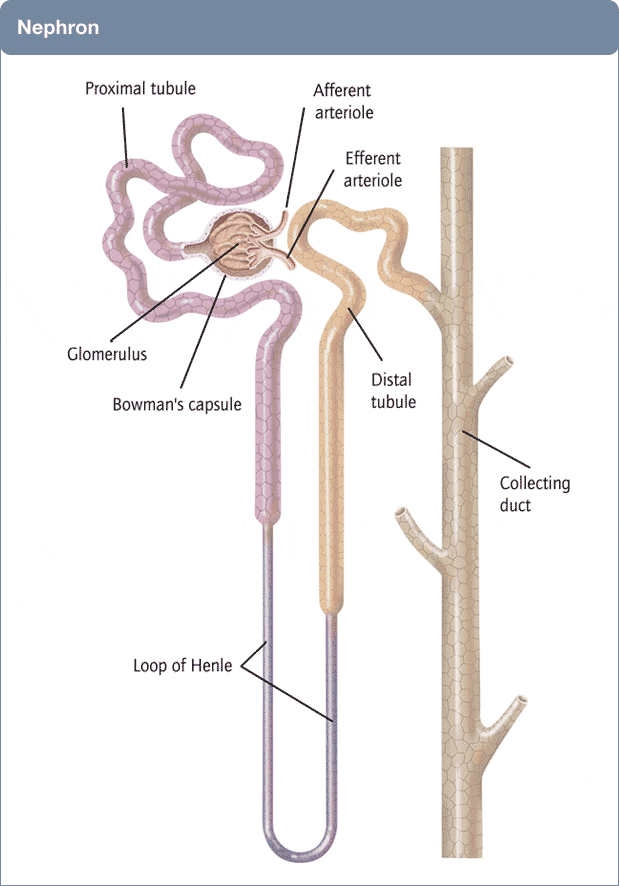 kidneys are made of about one million individual nephron units. The renal process begins with filtration of small molecules like citrate, or atoms like sodium and calcium, through the glomerulus, which is a complex of capillaries whose filtration pressure arises from the heart not a foot away.
kidneys are made of about one million individual nephron units. The renal process begins with filtration of small molecules like citrate, or atoms like sodium and calcium, through the glomerulus, which is a complex of capillaries whose filtration pressure arises from the heart not a foot away.
A majority of the filtered water, salts, and molecules is reabsorbed in the proximal tubule. The distal tubule (highly simplified here) performs tightly regulated absorption or secretion, so as to produce a final urine and maintain blood concentrations in their normal ranges.
These loops will come up again and again on this site so I should comment on the thin and thick portions. The long thin loops of Henle (Henle was the scientist who is credited with describing this part of the kidney) extract water specially well.The thick portions just below the ‘Distal tubule’ notation are called, appropriately enough, the Thick Ascending Limbs of the Loop of Henle. The thick limbs reabsorb NaCl, but not water, and in doing that entrain a marvelous system for – of all things – retaining water! In an article so long as this one, and concerned with citrate, I cannot pause longer here. But we will be back, someday.
Citrate is in the Blood
Kidneys Filter and Reabsorb Citrate
In one published study, concentration of citrate in blood is about 80 – 170 micromolar. A recent review places it at 120 micromoles/liter. If we use 120 micromoles/liter as a reasonable average, and a common value for glomerular filtration of 120 milliliters/minute, the filtration of citrate is about 21 millimoles a day. Of this about 1 – 4 millimoles appear in the urine, the rest being reabsorbed by the kidney cells. So the fraction of filtered citrate excreted is about 5 to 20%, and regulation of this fraction controls the amount of citrate in the urine.
Citrate in Blood Binds Calcium
The concentration in blood of calcium not bound with proteins is about 1 millimole/liter. Citrate concentration is about 0.12 mmol/liter, so in principle about 10% of non -protein bound – calcium can be bound by citrate. Because in calcium citrate crystals 2 citrate molecules can bind 3 calcium atoms, the the figure would seem to rise to to 15%. But in solutions like blood, other materials compete with calcium for a place on citrate – magnesium is one example. So the actual fraction is difficult to estimate. Normally blood citrate level is stable, so although significant, citrate binding of calcium is not likely to influence calcium metabolism by, for example, altering regulation of parathyroid hormone secretion.
Citrate has Signalling Roles
My purposes here are humble purposes, so all I wish to do is put here a tiny list of known effects of citrate on systems throughout the body without pursuing the details. Citrate concentration regulates lipid metabolism via malonyl-CoA. Citrate is sensed by the hypothalamus and thereby affects glucose intake and glucose metabolism by liver. To do these things citrate must enter the relevant cells, and it can do this only via a transporter that takes it across cell membranes.
The Citrate Transporters
NaDC1 and NaDC3
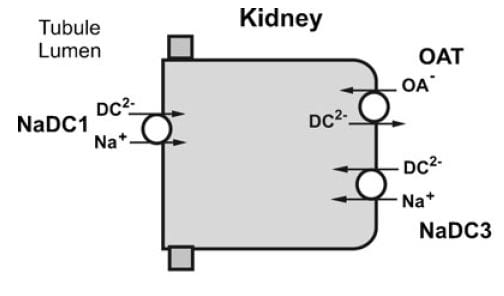
From Pfleugers Arch 466:119, 2014
NaDC1 is on the apical membranes of the proximal tubule cells of the kidney – the surface facing into the tubule fluid – and regulates the rate of reabsorption of the citrate that has been filtered. Its gene is named SLC13A2. This same transporter is on the food side of the small intestine cells and permits absorption of citrate from foods. The featured image for this article shows the structure of the transporter.
The citrate that enters the renal cells can be used for metabolism, or transported out the other side – called the basolateral side, facing the blood – via another transporter called the Organic Acid Transporter (OAT). Yet another transporter, NaDC3, permits citrate to enter kidney cells from blood. Because it appears to regulate urine citrate, my focus is on NaDC1.
The citrate transporter DC1 couples sodium and citrate movement. Since not everyone who reads this will know, let me mention an almost universal property of living cells: they pump sodium out of themselves and pump potassium in. Because they do this, sodium will tend to move into cells if given an opportunity – a hole. DC1 and DC3 can be thought of as sophisticated holes, or channels, through which sodium atoms can move if they have a citrate molecules with them. The actual proportions are 3 sodium atoms move with one citrate molecule, and the form of citrate which moves is one we have encountered before. Recall how citrate binds calcium because each molecule can have 2 or three negative charges on it. The doubly negative (divalent anionic) form of citrate is the one that traverse the channel.
They Transport More than Citrate
NaDC1 permits not only citrate to cross cell membranes but also succinate, alpha ketoglutarate, fumarate, malate, and a variety of less biologically relevant molecules. One might ask why, and I presume it is because the named molecules are all part of the citric acid cycle, which is the main engine of cell energy production. NaDC3 transports all of the same molecules as NaDC1, along with glutarate and a very long list of other molecules not in the citric acid cycle.
This cycle is at the center of that metabolism which uses oxygen to produce energy from food. The reference is to an excellent textbook review that is free online. Another chapter in that book finishes the story of how the cycle produces energy. The antiquity and centrality of the citric acid cycle will become apparent to you if you even browse these chapters. If you read them, you will encounter some of the most important aspects of living cells.
Why are Potassium Citrate Pills an Alkali Load?
In the citric acid cycle citrate is metabolized as citric acid, meaning that 3 protons are taken up from blood with each molecule. Removing protons is identical to adding alkali. Typical dosing is about 20 – 40 mEq of potassium salt daily, but the amount can vary widely.
Commercial potassium citrate contains 1080 mg of the compound in a 10 mEq pill. Typically the potassium citrate salts have a potassium on each of the three anion sites on the citrate molecule. The MW of citrate anion is 189.1. Urocit K, a common commercial version, is a crystalline monohydrate salt so it has a MW of 3×39 (for 3 potassium ions) + 189.1 (for citrate) + 18 for the one water molecule, or 324.1 in all. Given 324.1 for 3 mEq of base, the 10 mEq tablet contains 10/3 x 324.1 or 1080 mg.
The Flow of Citrate
In an earlier era organ physiology was popular and scientists often gathered together 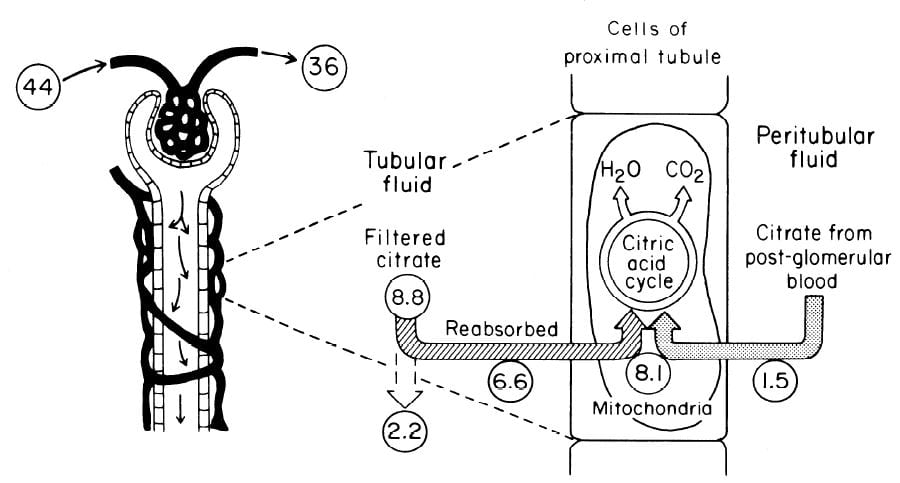 measurements to paint a picture of how things work overall. Here is such a picture from a wonderful review of renal citrate handling by Simpson. Values in small circles are micromoles (umol) per minute.
measurements to paint a picture of how things work overall. Here is such a picture from a wonderful review of renal citrate handling by Simpson. Values in small circles are micromoles (umol) per minute.
Citrate is presented to the glomerular filter at 44 umol/min, and 36 umol/min leaves the glomerulus (8.8 umol/min filtered) in blood what will pass by the blood side of the proximal tubules. From that 36 umol/min, 1.5 umol.min are taken up by renal proximal tubule cells and metabolized in the citric acid cycle. Of the 8.8 umol/min filtered, 6.6 umol/min are taken up on the urine side of the same cells making 8.1 umol/minute for metabolism. The remaining 2 umol/minute (3.17 mmol/day) are lost in the urine. NaDC1 and NaDC3 had not been cloned and sequenced at this early time, but physiologists knew the transporters were there and toted up what they did.
Urine Citrate Varies With Acid Base Status
Acid loads, such as high protein diets, will increase citrate uptake into the renal cells and thereby reduce urine citrate. Alkali loads such as diets high in fruits and vegetables or potassium alkali supplements reduce uptake and increase urine citrate.
Alkali
Clinical Response
In a trial, calcium stone formers with low urine citrate excretion eating a constant diet were given sodium bicarbonate or  potassium citrate, 20 mEq three times a day. Urine citrate rose with both treatments, as did the urine pH. Not relevant here, but in later articles, the sodium alkali did not change urine calcium, but the potassium alkali lowered urine calcium. Alkali itself lowers urine calcium, sodium raises it, and their antagonism is the reason for the differences.
potassium citrate, 20 mEq three times a day. Urine citrate rose with both treatments, as did the urine pH. Not relevant here, but in later articles, the sodium alkali did not change urine calcium, but the potassium alkali lowered urine calcium. Alkali itself lowers urine calcium, sodium raises it, and their antagonism is the reason for the differences.
Mechanism May be Increase of pH
If the citrate transporter is placed into test cells, the movement of citrate can be studied, and such a study shows how powerful is the effect of pH.
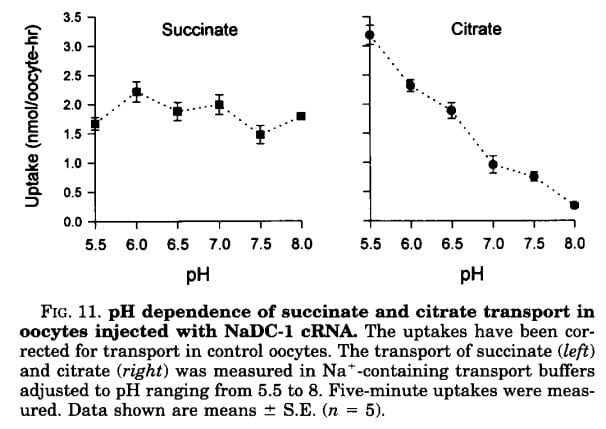 Succinate is a citric acid cycle intermediate like citrate, but its uptake by the citrate transporter is not affected by the acidity or alkalinity of the medium (pH). Citrate uptake is powerfully affected.
Succinate is a citric acid cycle intermediate like citrate, but its uptake by the citrate transporter is not affected by the acidity or alkalinity of the medium (pH). Citrate uptake is powerfully affected.
We have encountered pH before and remind ourselves here that urine values vary from about 4.5 to just below 8. Likewise, citrate has three sites that can accept protons, the acid component of water systems. As I mentioned in the paragraphs just above this point, the charge on the citrate molecule rises with pH as protons are progressively removed, and the sequence of pH values (the pKa values for the dissociating sites for those of you who know about such matters) are 3.13, 4.76, and 6.40. Obviously, in urine, the divalent (2 open negative sites) form will predominate until urine pH rises above 6 and will fall to about 1/2 of the total at 6.4. At about 6.4 transport of citrate was indeed just about half of that at the lowest pH.
pH in the Proximal Tubule
But it is not urine pH which affects citrate transport, it is the pH of filtrate in the proximal tubule of the kidneys, and that pH is not the same as that of the urine. At the end of the proximal tubule, the pH is about 6.7 to 6.8, and at that pH more than half of citrate is in the trivalent form and not available for transport. With alkali loads, as in the experiment in the table, the pH will rise, and citrate transport fall below normal, so citrate appears in the urine.
Problems with the pH Idea
Strangely, modern sources do not mention an older literature which raises questions about this mechanism. Simpson, in an important review from late antiquity (1983), mentions that the drug acetazolamide, which raises pH inside the proximal tubule and lowers pH inside the renal cells raises urine citrate only slightly and at first, but shortly after administration urine citrate falls despite a continuously alkaline urine and presumably tubule fluid. This suggests that even a high tubule fluid pH is not enough to counter the effects of changes in pH within cells or perhaps the blood. So it is not only tubule fluid pH that matters, but perhaps the pH inside the renal proximal tubule cell.
Acid Loads
Those unfamiliar with the matter may not realize that the diet we eat in the US and most of the other first world countries imposes an acid load that must be excreted daily in the urine. So the urine citrate excretion we find in our clinics and in experiments on ‘normal’ diets are those consistent with an acid load. When we give potassium citrate or other alkali we often do little more than neutralize this acid load, yet urine citrate usually rises. Experiments about acid loads add to the diet acid an extra amount of acid.
Tubule Fluid pH
As for alkali loads, a lower proximal tubule fluid pH will increase the fraction of filtered citrate in the divalent form which is transported by NaDC1. The pH of the tubule fluid will fall with acid loads for several reasons. Acid loads – for example a high protein meal – are buffered on blood bicarbonate which lowers the concentration of bicarbonate, and therefore the pH of the filtrate. LIkewise, the tubule cells are stimulated to increase their reabsorption of filtered bicarbonate which further lowers pH. All of this implies that kidneys sense the acidity or alkalinity of the blood, which they surely do.
Transport Adaptation
Over time – many hours to days – the NaDC1 transporter and its gene (SLC13A2) increase 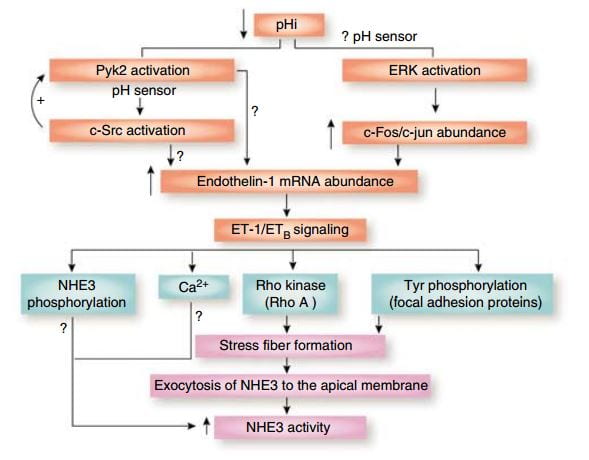 their abundances. This increase is mediated by endothelin – 1 (ET-1) through the endothelin B receptor (ETb).
their abundances. This increase is mediated by endothelin – 1 (ET-1) through the endothelin B receptor (ETb).
This figure from the above reference shows thinking about acid and endothelin as it was in 2007 and seems to be still. A fall in pH in proximal tubule cells can be sensed by a protein named Pyk2, which activates by adding a phosphate to one of its amino acids (tyrosine) and, interacting with another protein (c-Src), increases the abundance of the mRNA of ET – 1 which then signals through its ETb receptor to increase renal acid excretion – bicarbonate reabsorption – via NHE3, a transporter that reabsorbs sodium and secretes acid into the proximal tubule fluid.
This same ET -1 and its ETb receptor also signal increase of NaDC1 transport. Here, mice engineered to have (ETb+/+)or have not (ETb-/-) the receptor were challenged with an acid load. 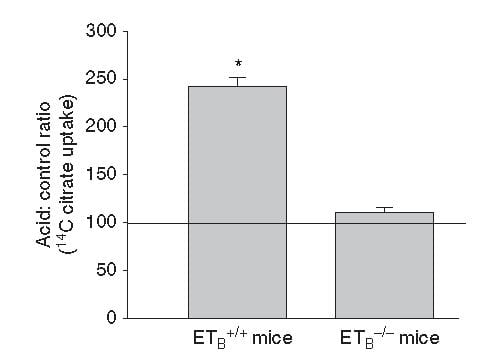 Citrate uptake by isolated NaDC1 transporters in the deficient mice do not respond to acid.
Citrate uptake by isolated NaDC1 transporters in the deficient mice do not respond to acid.
So one and the same effect, acid sensing and endothelin – 1 signalling increases acid excretion and citrate conservation.
But, you may ask, why am I grouping these two together?
It is because both concern acid base balance.
Citrate is metabolized as citric acid, taking up 3 protons per molecule metabolized, which is the same as saying it provides 3 molecules of alkali – like bicarbonate. Loss of citrate is therefore loss of potential alkali. NHE3 is a main driver of acid – protons – out of blood into proximal tubule fluid which reclaims filtered bicarbonate – conserving alkali.
So urine citrate, which we are interested in because it binds calcium and inhibits crystals, has a much larger role to play – part of the grand system which maintains a constant blood pH against the acid or base loads of diet.
Which pH?
I have spoken about pH of the proximal tubule fluid, of the blood, of the urine, but the one that is central to regulation of NaDC1 is the pH inside the proximal tubule cells. That pH appears to respond to acid or alkali loads, but the manner of its response is not simple. The signalling is through the Pyk-2 sensor already discussed and a parallel pathway via ERK (same diagram, above) which I did not discuss. But how sensing works, what is sensed, this remains very much an open research issues, and I will leave off here as this article was about urine citrate and the conversation has already taken us through many byways, beautiful if exhausting to follow.
Potassium
But – that awful word – one important fact remains to be uttered. Depletion of potassium lowers the pH inside kidney cells and lowers urine citrate. I will not pursue the details of this well worn story, except to point out its extreme clinical relevance. Diuretics that are used in stone prevention, or for hypertension, deplete cell potassium stores. It is the potassium citrate we give to patients.
Ammonium, and the Rest of the Story
How can I leave off without filling out the details of how kidney cells respond to acid challenge with production of ammonia that balances acid load with acid excretion?
Bicarbonate
A Better Buffer than Most
A buffer keeps pH relatively constant by taking up protons when they enter a solution and giving them up when alkali enters. It is a kind of shock absorber.
At the beginning, evolution favored bicarbonate. It is a buffer of considerable virtue in that it can take up protons or release them, like common buffers do, but has a special trait.
Bicarbonate is forever in equilibrium with carbon dioxide gas (CO2). When bicarbonate takes on a proton to become carbonic acid, much of that acid becomes carbon dioxide gas. When protons are taken out of blood, CO2 gas forms new carbonic acid which donates a new proton to the solution, and essentially bicarbonate appears in solution ‘out of thin air’. That it flows from solution into thin air and back makes bicarbonate a more stable buffer than those which live only in solution so it was an excellent choice.
What Kidneys do with Bicarbonate
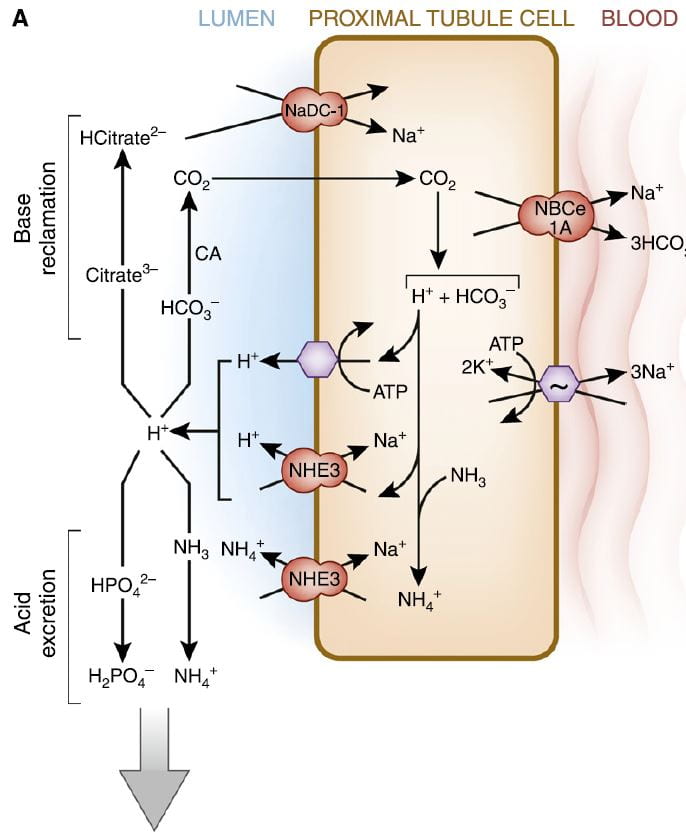 It is this very molecule, bicarbonate, which the kidneys traffic in when they respond to alkali or acid loads, and it is, of course, CO2 the lungs regulate in blood under the control of the brain.
It is this very molecule, bicarbonate, which the kidneys traffic in when they respond to alkali or acid loads, and it is, of course, CO2 the lungs regulate in blood under the control of the brain.
The figure is from the ‘A’ panel of a lovely drawing in a lively and engaging review. Being small, bicarbonate is filtered, and being the main buffer of the blood almost all of what is filtered must be reclaimed. So the proximal tubule cells, which do most of that reclamation, busy themselves forever with that task.
The way they do it is the simplest way. They add protons (H+) to bicarbonate in the tubule fluid, which becomes, as I have said, carbonic acid that transforms into carbon dioxide (CO2), which gas passes through the cell walls into the interior. Note, ‘CA’ is carbonic anhydrase an enzyme which speeds up the process of the transformation. In the cell, the CO2 becomes carbonic acid. Because protons are being pumped into the tubule fluid, protons are stripped off the carbonic acid so it becomes bicarbonate. The bicarbonate enters the blood with Na via the NBCe1A transporter.
There are two proton pumps. One uses ATP for energy to move the protons. The other (NHE3) uses the low Na in the cell as a gradient; sodium moves in through a channel like a revolving door, which makes one proton go out for every Na that moves in. At the blood side of the cell, the ancient ‘Great’ ATPase pumps Na out and potassium in, as it does in most cells that live on Earth. NHE3, the exchanger, is the molecule we met a few paragraphs above. It is increased by Endothelin 1 via the ET1b receptor.
At the top of the left side of the picture is citrate, our little slice of this massive structure. A few scraps of proton add to citrate so it has 2, not 3 negative sites, and can be reabsorbed. Its gene is regulated by endothelin 1 so when NHE3 is increased so is NaDC1.
Phosphate
Reclaiming bicarbonate is Sisyphean work. Nothing happens to get rid of acid loads from meals. But more protons are secreted than are needed to reclaim bicarbonate. Some are buffered on phosphate. But all the protons buffered on phosphate produce bicarbonate from carbonic acid inside the cell, and that bicarbonate enters the blood via NBCe1A.
Ammonium Ion
Ammonia is produced in the proximal tubule by removal of nitrogen from glutamine, pictured at left. As always, 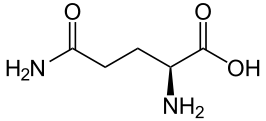 kinks are carbon atoms in this kind of drawing. The first one on the left has an oxygen and NH2 group, and a bond to the next carbon. Carbons typically form 4 bonds each. The next 2 carbons are merely linked to one another. The fourth has another NH2 and the final one at the right 2 oxygens. The left hand one is removed by an enzyme to produce NH3 and glutamic acid. The second one is removed to produce α-Ketogluteric acid which lacks any NH3. The 5 carbon skeleton remains unchanged.
kinks are carbon atoms in this kind of drawing. The first one on the left has an oxygen and NH2 group, and a bond to the next carbon. Carbons typically form 4 bonds each. The next 2 carbons are merely linked to one another. The fourth has another NH2 and the final one at the right 2 oxygens. The left hand one is removed by an enzyme to produce NH3 and glutamic acid. The second one is removed to produce α-Ketogluteric acid which lacks any NH3. The 5 carbon skeleton remains unchanged.
Ammonia (NH3) can tale up a proton to form NH4+, ammonium ion, which has a pKa of 9.3 meaning that at the pH of proximal tubules and cells, it is fully protonated. Loss of this ammonium ion in urine represents net acid excretion because the protons that were taken up came from carbonic acid which is converted to bicarbonate and transported into blood. Unlike titration of phosphate, excretion of ammonium ion does not increase urine pH because the pK is far above the pH of urine.
Under normal meal conditions, about 40 – 60 mmol/day of acid are excreted, of which about 2/3 is ammonium. Large acid loads, as for example, a ketogenic diet for weight loss, would induce a large increase in ammonia production so acid excretion can keep pace with acid production.
α-Ketogluterate
One might think this byproduct of glutamine metabolism, the 5 carbon skeleton, might be metabolized and done with, but no. A significant amount is metabolized. But some is not.
What is not metabolized traverses the kidney to cells in the later nephron, the intercalated cells in the collecting ducts, which usually pump protons into the tubule fluid to create the final urine p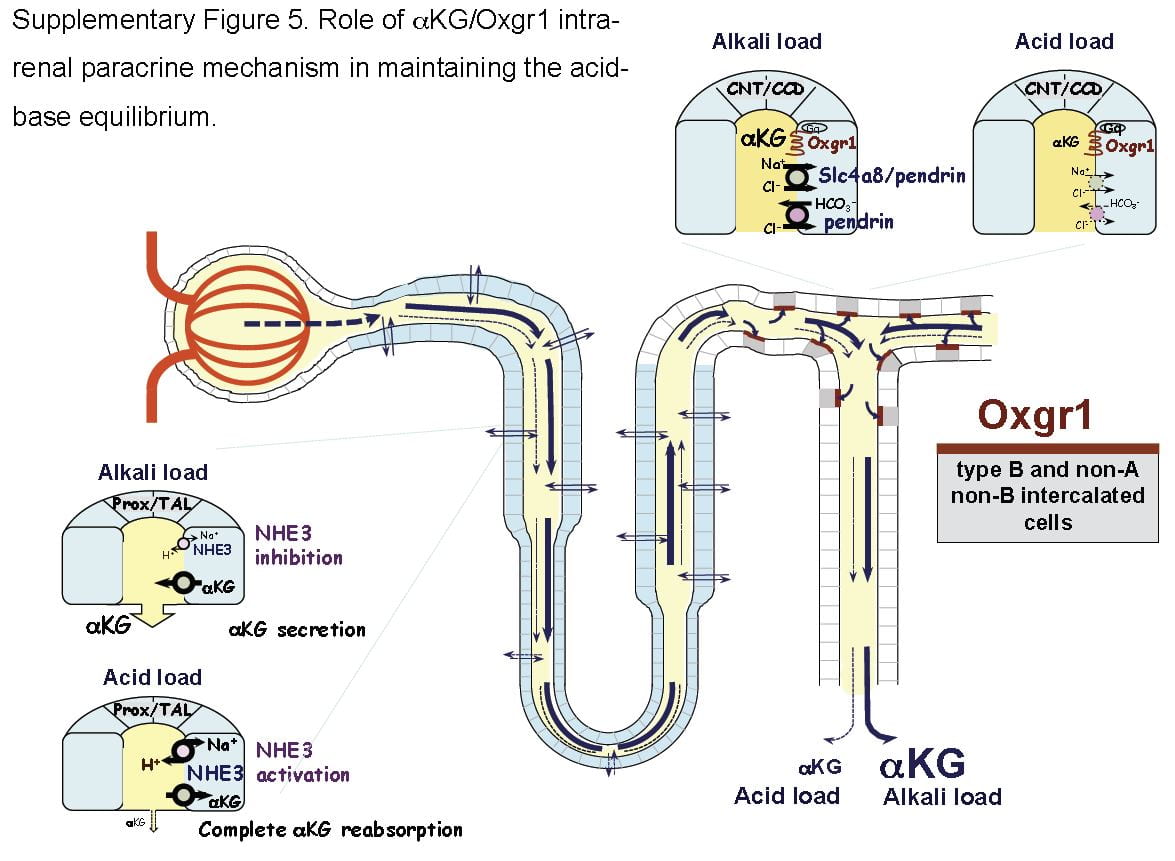 H which is critical to supersaturation and stone formation. But these same cells can reverse themselves and pump bicarbonate into the tubule fluid and protons into the blood, and they do this when confronted by an alkali load.
H which is critical to supersaturation and stone formation. But these same cells can reverse themselves and pump bicarbonate into the tubule fluid and protons into the blood, and they do this when confronted by an alkali load.
It turns out that α-Ketogluterate is itself filtered and reabsorbed in proximal tubule, and its reabsorption is profoundly reduced under alkali conditions so that more is delivered distally to a receptor (Oxgr1). When occupied by α-Ketogluterate this receptor signals the reversed intercalated cells (B and non-A cells) to increase their secretion of bicarbonate. The transporter for α-Ketogluterate is NaDC1. The net effect is to enhance bicarbonate – alkali – loss which offsets alkali loads.
The same receptor signalling stimulates pendrin, a complex exchanger which moves bicarbonate and Na together with chloride to effect NaCl and NaHCO3 reabsorption. Because acute acid challenge increases and acute base loading reduces proximal tubule NaCl reabsorption, this action would tend to maintain salt balance in that the intercalated cells would increase salt reabsorption as proximal tubule reduces salt reabsorption. Of note, although chronic acid challenge increases NHE3 abundance and activity, it reduces NaCl reabsorption via effects on other transporters. For these reasons the α-Ketogluterate – pendrin link is probably more important in minute to minute or hour to hour regulation than in adaptation to acid or base loading diets or treatments.
Citrate and Oxalate
You would think I had exhausted the topic by now, but no. NaDC1 and slc26a6, the citrate transporter and the anion 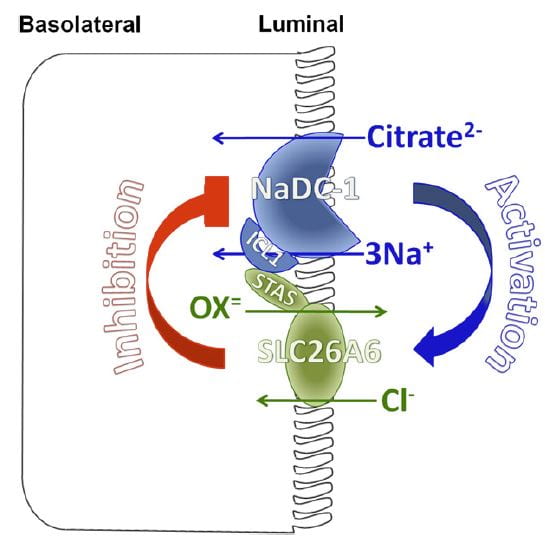 transporter (oxalate is an anion it can transport) which disengages NaCl transport from NHE3, themselves interact in relation to kidney stone formation.
transporter (oxalate is an anion it can transport) which disengages NaCl transport from NHE3, themselves interact in relation to kidney stone formation.
At least in animals and in cell experiments, the two transporters – which are present in a complex within the renal cell membrane – interact as in the figure. Slc26a6 inhibits NaDC1, so that when actively transporting oxalate into tubule fluid citrate reabsorption is reduced, urine citrate rises, and binds urine calcium to reduce risk of calcium oxalate stones. When oxalate secretion is minimal, NaDC1 increases to salvage citrate.
These animal and cell experiments imply that in human urine citrate and oxalate excretions should show parallel changes; this has not been tested.
Putting it All Together
Just Diet
Our urine citrate is an outcome of our biologies, which are variable, and our diets. Most of us eat a diet that imposes a net acid load, so our kidneys tend to conserve citrate and α-Ketogluterate, our intercalated cells pump protons not bicarbonate in to the final urine, our proximal tubules produce considerable ammonia and our urine pH is about 5 – 6.
Some of us, vegetarians whose diets do not have a proper balance of protein, very massive fruit eaters, as examples, have low citrate reabsorptions and high distal deliveries of α-Ketogluterate; our intercalated cells are reversed and stimulated to put bicarbonate into the final urine, our proximal tubules do not make much ammonia.
But the words ‘most’ and ‘some’ are misleading. In the US, certainly, chronic acid loading is the overwhelming rule, and the same throughout Europe and considerable parts of urban Asia. So our ‘normal’ poise centers on adaptations to acid load. It is not that we live in a neutral acid base condition, demanding from our kidneys little excretion of acid or of alkali. Life long we demand acid excretion. That is where we start. It is to that task our kidneys – and our bones, as I shall someday speak about – apply themselves all the days of our lives. However it is, for good or for evil, that lifelong adaptation to acid load affects us, that is our state, our permanent condition.
What Does Normal Mean?
When we give potassium citrate or any other alkali in doses of 40 to 60 mmol/day we neutralize a large fraction of diet acid. This is best considered not so much as an ‘alkali load’ as it is the removal of that acid load to which we have long been adapted.
Of course, urine citrate rises. Because we give alkali over months or even years, renal cells will adapt fully to the changes. But by ‘adapt’ I mean they give up the adaptations to acid loading. In the case where the dose of alkali just matches acid production one might best say the kidneys are relieved of their burdens in either direction, and reveal the way they would function if not driven to either extreme.
Like the small sailor plying the simple waters of a bay fills its sails sometimes southerly, sometimes northerly, making little way, dancing before a playful breeze, the cells shift their powerful machinery a bit here or there as one meal gives way to another. What shall I call this state of freedom? Why is this not the ‘normal’ from which point we register the responses to extra alkali or acid?
I have read where it was in Eden a condition of fruit, as the animals were not for them to eat. Perhaps I am wrong, and if Eden was as it says in our books the ‘normal’ state was alkali load. Perhaps Milton is wrong. After all he was not there, merely a poet making into life what he read in a holy book.
Potassium Citrate Pills
Raise Urine Citrate and pH
The expected changes are a decrease of proximal tubule reabsorption through reversal of the effects of chronic acid load. ET-1 signalling must fall, citrate reabsorption must fall because NaDC1 is no longer stimulated by ET-1 and because proximal tubule fluid pH will rise and with it the fraction of trivalent negative citrate.
Urine bicarbonate and urine pH will also rise. Partly, blood bicarbonate will rise and with it filtrate bicarbonate concentration and pH. NHE3 transport will be decreased vs. chronic diet acid loading, the baseline in the first world countries, and much of the proton secretion will be used in reclamation of bicarbonate. Naturally, NH3 production will be greatly reduced because the Pyk-2 sensing system will be signalling a higher pH.
Increases in Citrate and pH Vary Among People
But the biology is complex enough that in some people the main response will be citrate, and in other bicarbonate. Given all of the regulatory steps and signalling pathways involved a variety of responses is inevitable. Clinically this means one must measure and determine if the main effect is mainly increase of citrate excretion or of pH and therefore of CaP SS.
What is the Ideal Dose?
A nimble answer is enough to match net acid production – urine sulfate excretion is a decent index. I suspect that answer because of the problem of high urine pH in some people, and because as a clinician I never find it perfectly suits most patients. Yet it is a good starting dose because it aims at neutral acid base balance.
A Simple Pill with Powerful Effects
Physicians who treat kidney stones may well be the main ones who prescribe alkali loads to people with normal kidney function over months or even years or decades of life. This is indeed a remarkable physiological and clinical experiment, and that we do it makes the physiology and cell biology of acid base balance a central topic in clinical practice of stone prevention.
Likewise patients who take this humble medicine undergo what amounts to a reversal of cultural norm, which is a condition of chronic acid loading.
Thence, and for this reason, I have written a very long article about the topic, for physicians and their patients, and especially for scientists who know more about this topic than I do but may not see things from exactly the same view point.
Hello Dr Coe,
I just had a 6mm non obstructing Calcium Oxalate kidney stone removed from my ureter thru ureteroscopy. I have another 1mm stone in my other kidney. I had 2 24hr urine samples analyzed by litholink and below are the readings:
Day one/two results:
Calcium Oxalate Saturation: 2.27/2.02 Standard range 6-10
Calcium Phosphate Saturation: 0.14/0.10 Standard range 0.5-2.0
pH,24Hr,Urine: 5.664/5.594 Standard range 5.8-6.2
Citrate, Urine: 868/772 Standard range >450mg/24hr
Calcium, Urine: 172/144 Standard range <250mg/24hr
Uric Acid, Urine: 604/571 Standard range <800mg/24hr
Uric Acid Saturation: 0.61/0.65 Standard range 450mg/24hr) and starting Potassium Citrate raises this further will it cause any issues?
Concern 2) I read online in a medical journal that “Calcium oxalate supersaturation is independent of urine pH, but calcium phosphate supersaturation increases rapidly as urine pH rises from 6 to 7. Calcium oxalate stones form over an initial calcium phosphate layer….”
Will not increasing my Urine pH using potassium citrate not make me vulnerable to more Calcium Oxalate kidney stones?
Also, I read Urine pH <6, creates UA stones. Treated with alkali. My Uric Acid (Uric Acid, Urine: 604/571 Standard range <800mg/24hr). My stone was calcium oxalate, do I need potassium citrate?
Thank you
Viktor
Hi Victor, Your urine values do not confer risk of calcium oxalate stones. You left off urine oxalate, perhaps that is a problem. At 5 liters of urine volume you have no supersaturations so no stone risk. Since you formed calcium oxalate stones I deduce you have changed diet, fluids etc since having the stones removed, and therefore the labs cannot tell us why they formed. But you know what was present before, and I hope you will avoid that in the future. As for urine pH, it is not relevant for calcium oxalate stones even though they form on plaque. Before, when you were forming stones, I imagine your SS for CaP was above 1, which is more than enough. As for potassium citrate, in your present state why would you? Your urine citrate is high. Just do not go back to what you were. Regards, Fred Coe
Hi, Dr. Coe. Let me start by thanking you for all the informative help you’ve made available to the public in regards to kidney stones.
I am currently dealing with having passed my first stones 80% uric acid 20% oxalate. My 24 hr urine study showed my only problem was with 5.3 ph. I Had been on keto diet for 6 months prior to passing the stone (and still have one parked in other kidney) and really did not eat veges as I should have been, so mostly purines, Nor had i been a big hydrator (tho I am now (96oz a day), and Urologist says to immediately go on potassium citrate for life.
My question is this. Shouldn’t the doc have waited to see if I went back to a regular diet, with the 96 ounces of water I now drink, to see if the ph would balance out? And then, if ph didn’t rise, to then try a more alkaline diet with the 1/2 cup of lemon juice a day that I read is equal to daily doses of pharmacological therapy (I wont use the artificial sweetner aspartame they use in crystal light, tho I will use the crystal light “Pure” because the sweetner they use in there is stevia, but don’t know if the citric acid count is the same). to see if that would help first before putting me on a med for life?
Hi Jody, Given a uric acid stone you surely have the problem arising from low urine pH. Of course you should try higher urine volume and a more normal diet before committing to potassium citrate. If urine pH remains low on repeat testing, five servings of fruits and veggies is a good idea. If that is no good – and it may be inadequate – lemon juice is not my favorite. The fruit is too acid, and much of the citrate is in the form of citric acid that will not raise urine pH. Crystal light is reasonable. If you do not want it or it is not enough, perhaps you will need only 2 10 mEq K citrate pills a day, which is not bad. But do not leave the pH below 6 as the stones can grow rapidly. Regards, Fred Coe
Thank you for this very interesting article. I recently became interested in supplementing with potassium citrate after listening to a podcast interview by David Asprey (promoter of the Bulletproof ketogenic diet) with the recently deceased Dr. Richard L. Veech. In that interview, Dr. Veech stated that ketosis can precipitate gout because ketosis decreases elimination of uric acid, and Dr. Veech recommended that, if one was on the Keto diet, one should buy potassium citrate to allow the body to secrete uric acid. I recently was on the Keto diet and developed gout in the distal knuckle of a finger. Although I have no significant pain, the knuckle is quite swollen and red. I have no kidney stones that I know of. I have since switched to a more vegetable- and fruit-centric diet and greatly reduced foods with high purine content, but the gouty knuckle remains. I am wondering if that means I still have a high uric acid load and whether supplementing with potassium citrate might cure or reduce the gouty knuckle. Thank you again for your article.
Hi Pat, Measurement of serum uric acid is the easiest way to resolve the gout issue – if it was gout. One cannot know unless crystals were aspirated from the joint and identified as urates. A 24 hour urine will tell you if you produce too much uric acid. As for potassium citrate, it will not affect your present joint. Regards, Fred Coe
A month ago I had a CT abdominal scan for another issue (found to be clear) but some small kidney stones were seen, the largest of which is 3mm. I have no symptoms and have been drinking a lot of water every day since the finding, hoping to flush them out. The doctor says there is no need for treatment or follow-up unless symptoms emerge. I’d like to try to prevent any further formation and would be very grateful if I could be advised as to whether potassium citrate (or anything else) would be worth taking.
Hi Carol, I have to disagree with your physician – rather rare for me. If you indeed have multiple stones in your kidneys you should be evaluated as to their cause and treatment directed at the cause(s). This is my best summary of what to do. Regards, Fred Coe
The article was a great read but a little confused about the potassium citrate part still
Im currently experiencing oxalate dumping from leaving a 10 year plant based diet and reintroducing animal foods which have startup nutrients triggering calcium oxalate dumping, since I have been deprived for years the dumping is immense and depleting me of minerals very fast
On some advice I was told to take potassium and magnesium citrate alongside lemon juice daily to remove the oxalates from the body
But is your article suggesting potassium citrate depletes cellular potassium RBC?
would potassium chloride be better instead to replenish what’s Lost from oxalate dumping mineral depletion?
Hi Mathew, I do not have any evidence that oxalate dumping exists. The is no evidence of a link between oxalate and urine calcium and none between diet oxalate and other nutrients from a plant based diet. Potassium citrate cannot deplete potassium from red blood cells, potassium chloride has no known role concerning oxalate dumping. BEst, Fred Coe
Out of the blue, I had severe pain in my back on the left side. I’m a 53-year-old male that has never had kidney stones before, but, both my mother and father suffered from uric acid kidney stones. I have been on allopurinol for years for gout. I went to my general practitioner who took Xrays and a urine sample. While difficult to see, they identified two 3mm stones. My urine analysis found a PH of 6. Because of the coronavirus issues, my doctor did not recommend going in for a CT scan and recommended trying to pass them and he gave me a prescription of Flomax and a pain med. While we’re not 100% sure they are uric acid stones–my family history may point in that direction. My question is, with a PH of 6 would I benefit from potassium citrate supplements?
Hi Jon, Yes. The pH was in a single spot sample, whereas 24 hour urine pH may be a lot lower. But despite the virus you can get 24 hour urine testing which is done by mail. Litholink is running, and if your physician orders testing, they send out a collection kit and you mail back a sample. No virus exposure. Why not do that? Regards, Fred Coe
Thank you for this excellent article. I wish that this info was widely available to healthcare practitioners, as none of my doctors have suggested anything with regards to this intervention. As usual, I have had to rely on my own research to find the solution. I am diabetic (well controlled). I have gout and occasional kidney stones (usually small and easy to pass without much pain) and I have cut back significantly on meat and fructose. Cutting back on fructose is what made the biggest difference for me (the only fructose I was eating was in occasional honey, coconut sugar, and fresh/dried fruit).
But it’s not enough, because I just got a bout of kidney stones, the worst ever (I’ve peed out 6 in the last 48 hours and more are on the way). So it seems the next step for me is to hydrate WAY more, try to alkalize my urine as much as possible with diet, and also take potassium citrate. I also bought pH strips and will be monitoring my urine at home. I am also not getting a CT due to coronavirus unless the kidneys are shown to be irreversibly stressed.
Questions:
1) should I also be concerned about restricting foods high in oxalic acid? I know oxalic acid can also cause kidney stones in combination with calcium. Given that I also have gout, it’s likely that uric acid is the problem, but should I restrict oxalic acid rich foods just in case? I’ve avoided chard, spinach, and rhubarb for years ever since the first stone. I’ve also read that some stones are a combination of uric and oxalic acid?
2) I am concerned that so many stones have developed all at once. Is this explainable simply by chronic dehydration, high urine pH, and high uric acid level? I did eat more meat and homemade ketchup (high in fructose) than normal last week. Maybe it was a huge load of uric acid that totally overwhelmed the system. This episode has resulted in higher BP and kidney stress, but these are slowly resolving.
3) How long does it typically take for a uric acid stone of significant size to form? Days, weeks, months?
Thank you!
Hi Mica, Of highest importance is the nature of the stones. As a diabetic uric acid would be expected. Likewise urine pH would be low – below 5.5 in general. The fact of many stones all at once makes me think uric acid is the true cause. If it is uric acid stones, diet is insufficient and you require supplemental alkali. If the stones are calcium oxalate, perhaps oxalate may matter. If they are calcium phosphate oxalate is a minor thing. Get the stone analysed. A clue, often UA stones are red or red brown. Regards, Fred Coe
Hello Dr. Coe, Thank you for your article and education. I am diagnosed with early-stage ADPKD. I have been reading that potassium citrate and citric acid may slow the progression of cysts. I understand a trial is underway for the use of oxypurinol. If approved it still may prove unaffordable as a preventative treatment, and I’m not sure about the safety of using allopurinol long-term. All considered – my understanding is that tolvaptan may not have a major impact on renal preservation with ADPKD, so I am thinking to seek natural dietary intervention of alkalizers as my long-term treatment plan. Will discuss this further with my nephrologist but tolvaptan aside, a low-plant-based-protein and high water intake have been the advice so far.
Hi Andre, I went over your questions with Dr Arlene Chapman, who is my colleague at the University of Chicago and an expert clinician and scientist in the field of ADPKD. She offers her email address and a willingness to speak with you directly: Achapman1@medicine.bsd.uchicago.edu. I would take up her offer as she knows what you want to learn. If you choose rather otherwise, she is willing to tell me some answers to your questions that I can forward, but that seems to me a less ideal course of action. Regards, Fred Coe
Dr. Coe, That you took the time to make this connection is meaningful to me. Thank you for your kindness. I will contact Dr. Chapman, who is likewise so generous in her offer. My best wishes to you and yours.
Hi Dr. Coe, I have had kidney stones since 2002 and have taken potassium citrate pills daily to prevent them. I’ve increased drinking water daily, cut back on eating meat and fish, cut back on coffee, etc. In the past, my Urologist had me take Potassium Citrate 10 MEQ once a day, which helped. Then they had me see a Nephrologist who switched me to only 5 MEQ pill once a day, and in the past year alone, I had more kidney stones than ever. He won’t switch my dose back until my 24- UA collection, which I’m doing this weekend. My concern is, I’m trying to order “extra” pills incase we ever have an earthquake again (Anchorage had a big one Nov 2018), and I’m worried incase I get separated from home and can’t get access to my pills. I’d like to have extra Potassium Citrate supplements on hand with me. But when I tried to order on Amazon, there are SO many brands and variety of doses, so I was wondering what is the reliable place to get supplements at or a certain brand, if you had any in mind? I also bought Chanca Piedra, liquid form, from our whole food store and I only take it when I feel I have a bigger stone and need to pass it. It tastes awful after I dilute it with water, but it worked for me. Now, I’m just trying to make sure I have extra Potassium Citrate pills on hand, incase of emergencies. Thank you! Holly
Hi Holly, 5 mEq of potassium citrate is very low, and not a common dose. Even 10 mEq once a day likewise. I would speak with my physician about why so little. As for Amazon, all pills that contain potassium citrate OTC are low dose, about 3 mEq each. That is because of the potential dangers from potassium. The brands do not matter so much as the content of potassium citrate. But if your physician has limited your intake perhaps you have some medical condition that affects your tolerance for potassium. Since I know nothing about you, I can only suggest that you discuss the matter – of dose and Amazon – with your physician and understand what reasoning governs your present treatment. Regards, Fred Coe
Thank you Dr. Coe, both for this informative article and your willingness to interact directly with readers.
I first had a (calcium oxalate) kidney stone at age 62. I did not (knowingly) pass another stone until I was 67 (also calcium oxalate). At that time, a scan showed that my kidneys harbored perhaps a hundred stones of various sizes.
After my first scan, I was advised to follow a low-oxalate diet, take calcium citrate supplements and drink lots (!) of water. I did that, from age 62 on, but doing so did not stop the formation of many new stones. (I continued on a high protein diet during those years, which I had begun around age 59.)
My research led me to ask for a Potassium Citrate prescription; I now take 20 mEq daily. I am also on a low dose diuretic, to reduce the amount of calcium my body dumps into my urine.
I understand this regimen may help prevent FUTURE stones. However, is there anything more I can do to get rid of the stones I already have?
Thank you for your guidance and generosity.
Hi Pamela, No treatments dissolve calcium oxalate stones. But why so late in life?? Take a look at this article on mid life stones and be sure you do not have some special cause.
Dr. Coe,
I clicked on Jill Harris’ diet program site and received a letter stating, “Currently, the only type of stones that can be effectively treated with pills are uric acid stones. Potassium citrate pills can, in fact, dissolve this particular type of stone. However, uric acid stones account for only 5-10% of all kidney stones as the vast majority of stones are calcium oxalate. Calcium oxalate stones require a more holistic approach to treatment.” Based upon what I understand from your articles, would you agree with this, and add that potassium citrate may have an important role in inhibiting calcium oxalate kidney stones in that the citrate binds with calcium, thereby inhibiting calcium oxalate kidney stones; but that said, the best course is to first lower sodium and increase calcium, and then focus on the total amount of food oxalates consumed, and then, if necessary fit potassium citrate into the mix?
Really enjoy your articles!
Informative and fun!
Thanks
Roger Coggan
Hi Roger,
I am terribly sorry that perhaps my wording was taken out of context in my email. To be clear, I was simply stating that uric acid stones were the only stone that could be dissolved with the use of a pill, that being potassium citrate. But it is not its only use. Of course, it can be used as an effective treatment in regard to calcium oxalate stones as well. Many patients will ask if calcium oxalate stones can be dissolved and that was my point. They cannot, but uric acid stones can. I am sorry for any confusion.
Very best,
Jill
Hi Jill Harris I’m not a native English speaker and I wasn’t able to comprehend you fully. May ask you which of this statements is true:
[A] Potassium citrate can stop calcium oxalate stone from Forming and Growing but it could not Dissolve it.
[B] Potassium citrate can stop calcium oxalate stone form Forming but it couldn’t stop it from Growing and it couldn’t dissolved it either.
Thanks, Al
Hi Al, Potassium citrate can reduce new stone formation when used properly – here is a good article on its use. It cannot dissolve calcium stones but can dissolve uric acid stones. It may reduce growth of stone fragments left after stone surgery. Regards, Fred Coe
Hi Roger, Potassium citrate has been used in multiple trials against calcium stones with reasonable success, How it works is probably in part calcium binding, partly inhibition of crystallization. But in the linked article I do make clear that I try to take diet to the best point possible and then add citrate or thiazide as needed. Regards, Fred Coe
Hi Dr. Coe,
I was recently prescribed Potassium Citrate for Calcium Stones. I have a long history of stones and have endured several surgeries and the passing of stones. My most recent surgery was 1.5 years ago on my left kidney and I have stones there again. My urologist says I can expect to have surgery every few years to remove the larger stones in my kidney. She decided to try me on the Potassium Citrate. Do you think this will be helpful?
Hi Kristine, Stone prevention is organized – causes then treatment against the causes. Here is a decent overview of how to get evaluated and treated. See if it helps you in your situation. I am not a fan of just starting something by way of treatment. This is because a lot of stone risk arises from diet, and that can be changed for the better. Regards, Fred Coe
Hi Dr. Coe.
I got diagnosed with kidney stones after having my 4th child and only c-section. I had Problems 5 days post parteum with high blood pressure. Then a year later I had Urinary problems and they found a 20mm stone in right and 8mm stone in left. I had A PCNL for my right and during that they noticed i also had ureter stenosis in my right. and he wanted to watch my left 8mm stone. I fo Back 9 months later the 8mm stone grew to 17mm and I already Had 2 small stones measuring 5 and 3mm in my right where I had My previous surgery. At that time the dr put me on 2 5mg tablets of potassium citrate twice a day. So 25 mg a day. I can Only take the 5mg because those are the only round ones and I have Issues taking pills. My 24 hour urine test only showed a slight increase in calcium in urine. Not sure what that means and he said it was slight i don’t know the exact number. I went Back 4 months after having the 17mm removed via PCNL and he only saw 1 stone In My right at that time measuring 8mm. Which the other two were 5 and 3 mm so that equals 8mm and he thinks the stones fused together. Well I have Been on 25mg potassium citrate for a year now and recently I have Noticed the pills in my stool. They look completly in tact and not dissolved. What could be the reason for this. I asked The pharmacist and he said that should happen in those pills. I called My dr and I’m waiting to hear back from them. I just Find it odd after a year of taking them I find Them in my stools. Occasionally they upset my stomach and I get Diarrhea. I always Take them with a glass of milk and drink water after. I have To use milk because I need A thick liquid to take pills. What are your thoughts on why I coukd Be seeing them in my stool.
Hi Kate, I gather you had high blood pressure with your pregnancy. I hope that has been treated, and perhaps went away. It is very important blood pressure be controlled. Likewise right side drainage seems impaired by a stricture and stones have recurred there. On the left stones have grown. So you have active stone disease on a substrate of at least one episode of hypertension with pregnancy, something that can affect kidneys – albeit often quite benign. You do not say what your stones are made of, but possibly they are calcium phosphate, even brushite – a special form of calcium phosphate. You need to know. As for the pills, the wax matrix is in the stool but the medication has been absorbed. That is not your problem. Your problem is to prevent more stones, especially on the right where drainage is poor and a PERC had already been done. If your urine calcium is above 200 mg/d it needs to be lowered. Your evaluation needs to follow a plan. Regards, Fred Coe
Hi Dr. Coe,
I have been diagnosed with kidney stones I think 2 yrs ago. It was removed via ESWL. however after about 8-10months, it recurred. I was given Potassium Citrate to dissolve the stones. And it did.
Is it safe to have the potassium citrate can be used as maintenance pill to prevent kidney stones to recur. It seems that potassium citrate fits to dissolve the type of kidney stones I have.
I am afraid to have my kidney stones recur if I will just rely on diet (this is what I did after ESWL) as most of the food contains sodium.
Thank you.
Best Regards,
Prince
Hi Prince, That the stone dissolved means it was uric acid. Calcium oxalate and calcium phosphate stones will not dissolve with potassium citrate. If that is true you should be on potassium citrate long term to prevent more stones. Your physician has no doubt analysed some stone material, or measured the CT density of your stones – uric acid stones have low density. Regards, Fred Coe
Is there anything that dissolves and prevents calcium oxalate stones that you can recommend?
Hi Bre, The whole site is about prevention of stones like calcium oxalate stones. I am afraid you cannot dissolve them, however. Here is a good start on prevention. Regards, Fred Coe
I was hospitalized May 10th for critically high plasma potassium (7.0 MEQ/L). At the time I was taking a high dose (200mg daily) of a potassium sparing diuretic, Spironolactone, prescribed by my hepatology specialist to control Ascites. I was also taking Losartan Potassium (100mg daily), prescribed for hypertension by my Nephrologist and Potassium Citrate 10 mEq twice daily for kidney stones. When I was hospitalized, all of the above were stopped. Hypertension is controlled without the Losartan and I did not resume (at first) the potassium citrate. My ascites accumulation immediately returned at a rate of about 1 liter per day (10 liters removed by paracentesis every 10 days). My hepatologist started increasing my Spironolactone with weekly checks of my plasma potassium and I had reached 100 mg without increasing my potassium above normal 4.2 after one week at that dose and reducing the ascites to 3 liters in 10 days. I restarted the potassium citrate and within a week, my plasma potassium rose to 5.3. I’m not sure whether this was a delayed result of increasing the Spironolactone, a result of restarting the potassium citrate, or both. That background is to explain the urgency of my question for you. Is there another effective treatment for kidney stones that does not increase plasma potassium, i.e. something other than potassiun citrate?
Apologies, Dr. Coe, I left out a key bit of information. Analysis of a recent stone showed it was Uric Acid Dihydrate 80% Calcium Oxalate Monohydrate (Whewellite) 20% .
Hi Ken, Indeed. Uric acid does require alkali. Is your urine pH still low despite liver disease? Sodium alkali would not be reasonable with ascites. How about a thiazide like drug – chlorthalidone – or a loop diuretic might to help with potassium loss and fluid management, and perhaps allow a lower dose of spironolactone – diuretic drugs helps increase renal sodium and potassium losses, aldosterone blockers cause potassium retention. Your situation is complex, and I say this in ignorance of your situation, but perhaps single drug management of ascites is not ideal in that the potassium blockade prevents uric acid stone prevention. Regards, Fred Coe
Hi Ken, given the need for a blocker of aldosterone and one brush with serious high serum potassium, are your physicians sure you benefit from potassium citrate for stones? Are your stones still actively forming? What are they made of? Does your 24 hour urine still show significant stone risk? From what? I am rather skeptical, but far away and ignorant of your specific situation. If your stones are uric acid – a reason for alkali – can be find alternatives?? Regards, Fred Coe
Hi Dr. Coe.
I was diagnosed with Sandlike calculi in the mid calyx of Left kidney last July 3, 2020. My 1st Doctor prescribed Rowatinex 3x/day for 1 month. I started taking it for 13 caps already. I got to have 2nd opinion to another Doctor. And He prescribed bto continue Rowatinex and added Potassium Citrate 1080mg 2 caps BID. My serum uric acid was high 434umol/l. And he added Urinorm 40mg 1tab OD. My concern Doc. Is it okay to take K Citrate 1080mg 2caps BID? I Have Mild MVP with MR. Please reply Thank you.
Hi Anne, Your physicians are acting as if the sand is uric acid – I presume it has been analysed or is pink or orange/red. Rowatinex is useful only for passing a stone more easily. Urinorm is the trade name for a drug that reduced uric acid production. It will have no benefit for uric acid stones. Potassium citrate will raise urine pH and is the perfect treatment for uric acid stones. Is the sand uric acid? If the stone is still in the kidney your physician can measure its density on CT scan – very low in uric acid. Without more knowledge of your case, I cannot say more. Regards, Fred Coe
Hi Dr. Coe,
Thank you for your response. Is it okay to take K Citrate 1080mg 2caps BID? I am worried that the medicine will have an effect on my heart. And it makes my body weak and more sleepy. Is that a normal side effect? Thank you Doc.
Hi Anne, Your physician can measure serum potassium after you have taken the medication for a while, and be sure it is doing no harm. Sleepiness is not a known side effect of potassium citrate – not known to me at least. Regards, Fred Coe
Hi Doc,
I am a 62 yr old female, with several health issues who has been struggling with uric stones for several years. A Urologist some years ago told me that I have metabolic stone disease, but didn’t offer any treatment or solutions.
I’v been on Allopurinol 300mg for some years now, and still keep passing small stones on a fairly regular basis (especially following physical exertion). I also have a 3.5cm stone that bounces around inside, causing constant pressure and pain in my left flank and back.
Very recently another Urologist prescribed Potassium Citrate 1080mg (2 tabs × 3 times daily). I just started today. He also suggested a vegan diet.
What are your thoughts on this treatment regime?
Any hopes of my boulder dissolving?
Thanks,
Elana
Hi Elana, Potassium citrate is perfect for prevention of uric acid stones. Diet is irrelevent. Stones can dissolve, in part or entirely. Regards, Fred Coe
Hello Dr. Coe,
I was prescribed Effer-K 20mEq. It really irritates my stomach and I had to stop taking it. Can you recommend any alternatives that will not irritate my stomach. Thank you.
Hi Jim, the best approach is for your physician or pharmacist to offer a variety of products that provide the potassium citrate and you choose the best one for you. I find people all vary in their tolerances. Fred
Hello Dr. Coe, Thank you for your articles. I was prescribed Effer-K 20mEq, because of the low urine citrate in my Litholink 24 hr test, multiple non-obstructive small stones and increased creatinine. I took it for a while and citrates increased and creatinine lowered. Unfortunately it significantly irritates my stomach and I had to stop it and citrates decreased again. Can you suggest different medication/supplement that will not irritate my stomach as much. Thank you.
Hi Jim, I answered below. Take a look. Fred
Good Day, Doctor Coe:
I would greatly appreciate your feedback, please? I was hospitalized last autumn, 2019, for removal of multiple, renal calculi in the left AND right kidneys. I underwent four procedures and developed a severe hematuria infection and was hospitalized three-months. The stones from both kidneys were removed due to an excellent surgeon, but seven months later, I’ve developed a 4 mm stone in the left kidney that suffered the unforgettable hematuria.
My question is this: Is it safe to begin ingesting a smaller dose of Potasium Citrate daily to help pass this solitary stone AND prevent others. I have been ingesting citrate from lemons and extra virgen Olive Oil to help with the dissolving of the o-n-e 4 mm stone.
I truly appreciate your feedback……suggestions to be rendered…….advice after my very
unpleasant and prolonged hospital recovery.
Sincerely and great thanks for your response!
Dr. Don Contrer, DCN., MD.
Hi Doctor, You do not say, but are the stones uric acid, or is your 24 hour urine very low in citrate? The dose of alkali depends on the goals – urine pH, citrate and the stone. If you tell me the stone type I can do better. Best, Fred
Good Day, Sir:
My multiple renal calculi removed this past winter were termed: Oxalate stones. This solitary stone I have concerns me and my daily half-dose of Potasium Citrate. The dosage causes me heavy lethargy and tiredness.
How often must blood work be rendered and what are the specific areas one should be concerned?
Thank you for your RESPONSE.
Dr. Don Contrer, DCN., FAAD
Your response to my last comment I sent two weeks ago on August 15th, please?
Thank you,
Dr. Don
Hi, I did write – sorry to be slow, but right now things are so hectic! Fred
Hi Doctor, The common stone has many causes, and perhaps your treatment is best or not. See what you think about the article – my favorite – as to alternatives and whether you have had optimal evaluation. Best, Fred
Hi Doctor Coe.
Thank you for your article.
I’m a 61 yr old male, good health, in good shape, good diet. I’ve had a few stones over the years. 2 lithotripsy procedures. One successful, one not. All in all, kidney stones are not fun.
My Nephrologist prescribed Potassium Citrate 15MEQ twice daily. I need to follow up w a blood test in a month.
I’m not thrilled about taking a medication twice daily for possibly the rest of my life, especially one that requires regular blood testing.
I’m always concerned about adverse reactions to medications especially over long term.
We are not really “pill people” in our home. I’ve had back surgery and several dental procedures. I took pain relief for a day.
Can you please shed some light on my concerns in regard to long term usage of Potassium Citrate?
Thank you
Follow up;
Stones are reddish. Have to assume they are uric acid.
Wow, they are. Get them analysed!! Sometimes they are uric acid +. Fred
Hi Neil, a lot depends on what the stones are made of, and what the 24 hour urine samples show. For example, if urine pH is low and stones are uric acid, potassium citrate is crucial. IF stones are calcium based, perhaps urine citrate is very low. Perhaps the medication was given I would ask your physician why this medication is needed, and about its possible drawbacks. Regards, Fred Coe
Hello Dr. Coe.
Thank you for your article.
I’m a 61 yr old male, good health, good diet, in shape. Several stones, reddish in color, over the years. Starting about 35 yrs ago. 2 lithotripsy procedures. One successful, one not. Stones are not fun.
My Nephrologist prescribed Potassium Citrate 15MEQ twice a day.
We are not “pill people”. I’ve had back surgery and many dental procedures. A pain relief pill for maybe a day.
I have concerns in regard to a medication I need to take, possible for the rest of my life. Especially one that will require regular blood testing.
Can you please shed some light on long term usage of Potassium Citrate?
Thank you.
(I wrote earlier but it didn’t post)
Hi Neal, I think I answered this below. Fred
Hi Dr Coe,
For a patient on Chlorthalidone, which is a carbonic anhydrase inhibitor and causes the kidney to excrete Bicarbonate, would it be helpful to take a Potassium Citrate supplement? Or Potassium Bicarbonate? And why?
Thanks.
Hi Neil, The CA inhibition gives a small bicarbonate loss, and actually urine pH falls. But volume depletion from the diuretic + continued distal delivery because of blockade of the DCT NaCl cotransporter creates potassium depletion. That in turn increases PCT citrate reabsorption via NaDC1. I usually use KCl, cheaper, and as potassium stores rise urine citrate will rise as well. I take it you are a physician. Best, Fred
Thank-you Doctor Coe. Your brilliant analysis of potassium citrate usage was exactly what I was searching for and your astute classic literature references humanized my desperate search. I have been prescribed 6 months of k-citra 1080 mg take one or two tablets twice daily. I have rarely taken an aspirin. December 2019 I went to emergency with blood in my urine.I am 64 years old in good health although some might think i am obese 5foot 8 and 275lbs . I lifted 8tons of steel 5days a week for 30 years. Gained 100lbs when I quit smoking 12 years ago. I was diagnosed with bladder stones. The picture I saw from the stirrups looked like three strings of golf balls my surgeon counted 15 too large to be removed thru the penis. Dr Troy Schultz removed between 2-3 cups of stones from my bladder(amid lots of oohs and ahs from the excellent support staff) Recovery seems to be going well, my follow up appointment with Dr Carole Connick. announced that my stones contain Gout! Yikes old grampa had a heck of a time with his touch of gout and my feet are extremely sensitive and cramp up sometimes when I put on my socks. My doctor was mindful of my natural existence and let me make the choice between” Dilution being the Solution” to quote Harvard Medical or taking potassium Citrate. I drank beer like a fish for 40 years, but I was also a provincial champion Speed Skater at aged 28. I quit alchohol nicotine but I still chase my wife around the house. My dad is 88 and in fine shape, my mother is 86 and has suffered the ravages of chrones disease her entire adult life. I’ve taken up way too much of your valuable time just by reading this ,my main goal was to thank-you for this article and any Canadian of average intelligence can make up one’s mind having been provided such eloquent information. Having taken up so much of your time I would not be hurt by no response but would be thrilled by a one word or possibly a very succinct phrase to sum up Thanks again (in good hands) GFJ
Hi Guy, The stones being gout’ means – I believe – uric acid, and there is no choice: raise the 24 hour urine pH above 6 and they will never come back. No amount of water is as good. But be sure to get 24 hour urine testing so you know the dose of the k citrate is adequate. Regards, Fred Coe
Hi I just read your article I had 8 big stones blasted August 25th was put on Potassium citrates ER 10 meq 3 tablets 2 times a day. If I just take 1 two times a day will it help me. I am having problems taking more of them.
Hi Shirley, Treatment depends on the cause of stones, so be sure you have been properly evaluated with 24 hour urine testing and serums as well. Potassium citrate may or may not be useful for you depending on the results. Regards, Fred Coe
I have had two stones – uric acid (7 mm and 5 mm) in the past 8 months. Both removed with in the hospital. I am now on Allopurinol. I have friends with similar problems and all take K citrate. My urologist says he does not know if one is more effective than another but always prescribes Allopurinol. I cannot find any efficacy studies of the two with uric acid stones. He says I can try potassium citrate for a month and see how I do. Is there a reason, other than compliance due to one pill of Allopurinol versus Urocrit three times a day, to take one over the other. I would really appreciate an opinion. Have 4 or 5 small 1 – 3 mm renal stones at present.
Hi Martin, There will not likely be a trial because urine pH so much outweighs urine 24 hour total urate excretion allopurinol cannot possibly match alkali such as potassium citrate. Low urine pH is THE cause of uric acid stones – if you want real detail, here is the technical chapter. Use the K citrate, aim for 24 hour urine pH >6. Regards, Fred Coe
I have uric acid stones. Otherwise, I am in good health. Doctor has me on Allopurinol. I have a number of friends on Potassium Citrate. I cannot find anyone who tell me if one is generally more effective than the other. I do not care about the convenience, just the efficacy.
Hi Martin, I believe I answered this already. Fred
Hello Dr. Coe.
Thank you for your article.
My question is, in the treatment for urinary tract infection, how the use of potassium citrate would help to treat the condition? Can you explain in terms of the drug pharmacology and chemistry aspects?
Thank you so much.
Hi Lia, Potassium citrate has no role in treating urinary infection. Regards, and apologies for so late a reply – your question was lost. Fred Coe
Hello Dr. Coe. Thank you for your work on this matter. I am a live long producer of calcium oxalate kidney stones (over 40 so far). I was recently prescribed Potassium Citrate by my urologist. Should Potassium Citrate be as effective with calcium oxalate stone prevention as with other types of stones? Also, how long has the prevention of kidney stones being treated with Potassium Citrate? Thank you.
Carlos
Hi Carlos, Here is my article on treatment of your kind of stone. The trials are in it along with what you might want to do to be sure what category of patient you fit. Regards, Fred Coe
Hi Dr. Coe,
I am a calcium oxalate stone former (idiopathic), 39 y/o male. I know that the general recommendation is to take potassium citrate on even intervals – every ~8 hours. My urine pH is actually right about where it should be without it. So, taking just 2 x 10meq daily has given me 24 hour measured pH of 6.5. My citrate is just within normal (low side), I think in part due to my thiazide medication that has brought it down from my baseline closer to 500. Since I am unable to stay hydrated as well throughout the night, would it be beneficial to take 10meq K-Cit right before bed? Would that mitigate stone formation overnight in a way that is perhaps worth the risk of elevating pH temporarily? To put it more simply (which is maybe unfair): is citrate more important than pH to keep within range?
Thanks for all you do!
-Tad
Hi Tad, If your citrate fell because of potassium loss from thiazide – a common thing – potassium chloride would be the more reasonable replacement as potassium will restore losses and bring urine citrate back to baseline without the disadvantage of raising urine pH, or the cost of K citrate. Likewise, most find it easier to swallow. The timing is irrelevant. You are using K citrate to replete potassium losses, not as a primary stone prevention, but the chloride form will do as well. Regards, Fred Coe
Dear Dr. Coe, I underwent ESWL last October to get rid of two calcium oxalate stones, and this was the third time in my life (I’m 56). The stones, originally 13 and 9 millimetres, were finally expelled with the help of a stent in the ureter.
Now of course I am balancing my diet, drinking a lot more water than usual, and taking about 3.5 grams of K-citrate in two doses daily, morning and evening. In your opinion, how long in my life should I continue to take this salt? Thank you so much for your work.
Hi Renato, I do not know if potassium citrate is right for you or not given no information about your test results. Here is a good general introduction. See if you have been fully evaluated and if your results point to citrate as your best treatment. As for length, improved diet is best life long – or until science gives us something new and better. For meds, if needed, long term is my best comment – no one has any real data on ‘how long is enough’. If pushed I would say 10 years minimum – purely arbitrary and indefensible as an answer. Best, Fred Coe
Hi Dr. Coe, I have been reading your articles about potassium citrate and this line caught my eye: Citrate is sensed by the hypothalamus and thereby affects glucose intake and glucose metabolism by liver. My children and I suffer from non-diabetic hypoglycemia (no kidney stones). 10 meq x2/day of potassium citrate is like flipping a switch for me (symptoms improve dramatically), but no one can explain why (also won’t prescribe potassium citrate to my children). WES genetic testing at Johns Hopkins revealed a VUS in ATP8, but nothing else (we were put on a mitochondrial cocktail by a metabolic nutritionist which also helps somewhat). No one has any answers and my children and I suffer from neuroglycopenic symptoms daily, some of us also have a bad urinary odor (although this gets better with less protein, potassium citrate, high potassium diet and medium chain fatty acids seem to help as well). I am desperate for help at this point. Wondering if you could provide any insight as to the effects of potassium citrate and its effects on non-diabetic hypoglycemia. An additional note, I grew up in Europe where we did not drink still water but rather mineral water high in bicarbonate (Gerolsteiner), wondering if there could be a connection as to why these symptoms were not as pronounced in me as a child.
Hi cg, The non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS) is rare, and seems poorly understood. I have not read about citrate as a treatment. Citrate is a nutrient, at the beginning of the Citric acid cycle, so sensing is not a surprise. It is utterly harmless, and can be bought as a food additive as the potassium salt. You might mention this to your physicians, and if it is not deemed unsafe, use it. I have no specialized knowledge of this condition. Regards, Fred Coe
Dr Coe
Thank you very much for your insight. I will discuss with our doctors, just one more question. Does potassium citrate skew results looking for acid disorders/acidemias? I’m asking because I was taking potassium citrate when these types of tests were conducted and it seems to me from what I’ve read that potassium citrate should have been stopped prior to running the tests.
Thank you
Hi Cg, to get baseline data on what may be wrong, test without the alkali supplements, of course. If you need supplements, and are testing to see if the dose is proper, test while taking the supplement. Regards, Fred Coe
Hello,
Very interesting article, I would like to ask you a question which I did not find the answer to during the reading.
What is the timing of the peak concentration at muscle and blood level concerning potassium citrate?
Is it the same as for sodium citrate or magnesium?
Thank you in advance
Hi Lucas, In a normal person, the potassium is taken up into cells rapidly enough you will not see an increase apart from a research setting. Given the commercial pills are slow release, they dictate the kinetics – slow. Citrate is metabolized as citric acid in the liver via the Citric Acid cycle, producing alkali as the citrate takes up a proton from blood buffers. That is rapid but the kinetics are controlled by the pill release characteristics. Sodium citrate is usually a liquid, blood sodium cannot change at all from an oral sodium load, and I presume citrate in blood rises over perhaps 10 – 20 minutes or so – I have some preliminary data suggesting that. I know nothing about magnesium citrate. Regards, Fred
Hello Dr. Coe
I was wondering if potassium citrate had the potential to lower stomach acid (and therefore cause digestive issues and dysbiosis down the line).
Thank you!
Hi Myriam, No. The material cannot lower stomach acid because the citrate is not an alkali, it is metabolized by the body cells to bicarbonate but not by the stomach. Regards, Fred Coe
Dear Dr. Coe
Thank you for your articles. Generosity.
Are you able to make a guess here? My partner had a horrible kidney stone. We were given morphine and a funnel with a screen on it to take home in order to catch the stone. A vicious looking little stone came out. We let it dry out. I thought a stone would be hard. But when I put my fingers in to retrieve it with almost no pressure, the whole thing turned to dust and fell into the shag rug. There was nothing left to take to the Doctor. Does this give you a clue as to which type of stone it might have been?
Mike
Hi Mike, It may be a protein aggregate with only microscopic amounts of mineral in it. I would have submitted it for analysis as modern instruments might have detected the crystal structure. Other possibilities exist. I am sure the physician responsible will want to do a CT scan to look for stones, and do a standard evaluation to look for stone causes. There are other non stone causes the CT may reveal. Regards, Fred Coe
Hello Dr. Coe,
I suffer from kidney stone disease secondary to idiopathic hypercalciuria. I was really hoping to be able to use potassium citrate to help prevent future stones, but that might not be possible seeing as my urin pH is naturally between 6.5 and 7. Would it be possible to counteract the rise in pH due to taking citrate by taking some sort of acid supplement as a buffer? My limited understanding makes me believe that this would keep my urine calcium and pH where they are while increasing my urine citrate, but if it was this easy, surely someone smarter than me would have thought of it already! If it wouldn’t work, could you explain why? Thanks!
Hi Andrew, Potassium citrate raises urine citrate because metabolized as citric acid, thereby producing new bicarbonate alkali, so raising pH is inevitable. Adding an acid will merely undo the pill and maintain urine citrate where it is. Given IH and high urine pH, I would not generally want added alkali, but rather low sodium diet + a thiazide diuretic (that will lower urine calcium and also lower urine pH a bit). Regards, Fred Coe
Hi, some products contain potassium citrate and citric acid. what’s the role of the citric acid in the solution?
Does it complement the potassium citrate by increasing the filtered citrate? I understand it does not affect the Ph.
Can the clinical results be different than the ones obtained by only taking potassium citrate pills?
Hi Loren, Citric acid is citrate with protons on its negative oxygen sites. Being an acid, citric acid gives up its protons into solution readily leaving behind the citrate anion (negatively charged molecule). Citrate is metabolized as citric acid, and that means it takes protons up from blood, which produced bicarbonate – alkali. That alkali signals kidney cells to release citrate into the urine instead of metabolizing it for energy. Citric acid is just that, eat it and cells use it as nutrient, and kidneys excrete it – no value for stones. As for foods and beverages, one adjusts the ratio of citrate to citric acid by adjusting the pH (acidity) of the product to achieve the desired taste. Regards, Fred Coe
Dr. Coe,
Great article. I have a question though. It is implied that metabolism of citrate is “alkalinizing”. When (counting protons) citrate is metabolized to oxaloacetate via the TCA cycle the three NAD-dependent dehydrogenases lead to production of three protons for each citrate molecule cycled. To make one glucose molecule from part of the carbon skeleton of citrate would take two turns of the cycle (to get two oxaloacetate molecules to make one glucose molecule) and therefore would produce 6 protons. This would be acidifying. The final product of the cycle, oxaloacetate, can be used for gluconeogenesis and a proton can be consumed in the gluconeogenesis reverse reaction catalyzed by glyceralde 3-P dehydrogenase. Two of the latter reactions would be needed to make one glucose molecule. That still leaves six protons produced in the TCA cycle and two consumed for the two oxaloacetate molecules converted to glucose. Therefore, net production is four protons. What am I missing here? Thanks in advance your consideration.
Ray Corona
Nephrologist
Thanks, Ray, it is that food citrate is metabolized as citric acid, so it takes protons off blood buffers as it enters the cells. The buffer system then makes more bicarbonate. As for the overall metabolism of citrate, net proton production is not easy to quantify as the glucose system – as an example – links to fatty acid synthesis etc. Since citrate has three anion sites and all must take up protons, and diet citrate has no protons on it, there are 3 per mole taken up reducing the differences you mention. I appreciate your insight and that you for it. I did not explore the final fates of citrate metabolism in my article even though they are available. Best, Fred
What is the difference between Magnesium Citrate (MagOx) and Potassium Citrate? I see both being recommended for the prevention of kidney stones. Magnesium seems to have less side effects, but most providers prescribe potassium. Is this just two different options for getting citrate into your system/diet?
Hi Jeffery, I need to write about the various newer forms – so your question is right. You cannot tolerate too much magnesium citrate but it is a good part of a mix – magnesium, sodium, potassium citrate. Potassium citrate is usable straight, and, frankly, if a proper dose form were OTC nothing could match it for simplicity. But FDA worries about potassium loading OTC because some people have significant kidney failure or other rarer problems and cannot tolerate the potassium. Pricing for potassium citrate is outrageous. Thence the market for all of the other forms. Regards, Fred Coe
Hi Dr.
Would it be a bad idea to drink a solution of apple cider vinegar and potassium citrate together ?How about solution of magnesium citrate and potassium citrate? thanks
Hi Lisa, Here is the idea. First, do you need potassium citrate? As the article notes, it is of value but not always. Is your urine citrate low? Urine pH, is it low? If not, perhaps this is not going to be effective as a stone prevention. As for home brews, I do not know what apple cider vinegar with potassium citrate in it would taste like, and am afraid to find out – I cannot imagine a medical danger. Magnesium citrate is large amounts causes GI intolerance, and is not used in stone prevention to any important extent. Regards, Fred Coe
potassium citrate, alkaline citrate(moonstone drink) chanca piedra and hydroxycitrate along with lemon juice lime juice and apple cider vinegar. I utilize all of this with almost a gallon of water a day to try and dissolve a 9 mm stone. Do you think this will be effective?
Hi Patricia, all of these are potential sources of alkali that should in general raise urine pH and possibly urine citrate. I do not think calcium oxalate stones will dissolve, but uric acid stones could. Calcium phosphate stones might grow larger. Regards, Fred Coe
Patricia, I just had a lithotripsy procedure for a 7mm stone. It pounded the stone down to passable sizes. Good luck.
Hello Dr. Coe,
I have been prescribed Potassium Citrate (10 MEQ) twice daily for my chronic Kidney Stone condition. I also suffer from recurring canker sores. Do you think the acid of the citrate could be making the canker sores worse? Or can it cause stomach problems? I also have acid reflux.
Thank you,
Ali
Hi Ali, potassium citrate can surely worsen GI symptoms. I know of no link to canker sores. Regards, Fred Coe
I am 76YO woman. Oxalate stones since I was 25. Bilateral medullary sponge kidneys. Urologist just prescribed 10 Meq potassium citrate up from 300 mg/day. My potassium on recent blood test is 4.
I am concerned the higher amount of potassium dosage will cause too high blood potassium.
I just received first order of Moonstone. Will this add too much also?
I live in rural NJ and have access to one Urologist. He does not believe in explaining his treatments. I am a retired Histologist who constantly researches new methods. Just want some indication that 10 Meq is not too high a dose.
Hi Theresa, Moonstone is just another version of alkali, and the K citrate is enough by itself. If you have concerns, a fasting (pre-dosing) blood sample at one week of K citrate will disclose any problem with potassium. I would say that 10 mEq per day of K citrate is so low as to be unlikely to raise urine citrate – the only known reason to use the agent, and that 20 mEq/d is what I would consider a minimal dose. Regards, Fred Coe
Thank you for this excellent article! I seem to be one of the lucky ones in that I have had success with increasing my urine pH from 5.0 to between 6.0 and 6.5 by taking just 620 mg of potassium citrate per day, along with ~2,700 mg of magnesium citrate per day, in hopes of lowering my risk for more calcium oxalate stones. However, that only works during the day. When I test my urine just after waking in the morning, the pH test square remains stubbornly orange. I have sleep apnea, for which I use a CPAP machine. (I’m a 60 yo male.) My question is, should I be worried that a dip in pH at night will significantly impede the protective benefit I’m getting from the supplements—or is the 8 hours during which I’m sleeping at night a short enough period to be negligible?
Hi Jay, For uric acid, the urine pH is prime. For calcium oxalate stones the urine citrate itself is. So you want to know the 24 hour urine citrate – is it in a range above stone risk. In your case, calcium oxalate stones, urine citrate is what matters. Regards, Fred Coe
Thank you so much!
In my comment from earlier today, can you please change “Jay Kissel” to “Jay K” and change “mitigate” to “impede” before publishing?
Thank you!
Jay K
Hi Jay, I edited the comment but could not alter the personal header – not allowed as it comes from you. Fred
Thank you! No problem about leaving my full name. Please feel free to delete my request for edits.
BTW that’s a photo of my wife next to my comment, not me. It seems to have been appended automatically because she also has a blog using the same online service. Funny! JK
I noticed. Your computer knows what it wants to do. Fred
Wonderful, informative article. I have a genetic mutation which interferes with the reabsorption of Phosphorus. Over the past 10 years, I tried Low protein, low Na, Low sugar, low oxylate diets yet continue to have Hypercalcemia, hypercalcuria and calcium kidney stones requiring surgical removal about ever 2 years. My doctor recently prescribed KCL 30 mmol 2 times a day. I would love your feedback on this platform or you may forward any questions or information to my email address.
Thank you
Hi Marie, I gather you have high serum and urine calcium and calcium stones from an inherited disorder of phosphate transport. A majority of low serum phosphate phosphate wasting genetic disorders have normal serum calcium. A similar review found much the same. HHRH is the usual reason for stones in inherited phosphate transport disorders, but serum calcium is generally not high. Is it possible that you have primary hyperparathyroidism? The only reasonable inherited disorder is HHRH and serum calcium can be elevated in childhood but in adults is usually normal. This case (a lot like the other one I referenced) showed high serum calcium that faded by adult life. If indeed genetic analysis has proven you have the transport protein defect, I subside in bringing up primary hyperparathyroidism. Your treatment, however, seems odd in that phosphate supplementation is almost always needed in HHRH and helps to reduce the high 1.25D levels that drive high urine calcium. Diet changes you note will not be effective in HHRH. Regards, Fred Coe
Hi Dr. Coe! Per my August, 2021 24-hour urine test results, I’ve been taking 25mg of HCTZ (1/2 pill in AM, 1/2 in PM). My January, 2022 24-hour results are in (see below), and my urinary calcium has dropped nicely, but so did my citrate levels. My diet has not changed, in which I continue to be ~90% vegetarian, getting my ~1,200 calcium via vegetables and dairy. I also drink 1/2 cup of lemon juice mixed with a gallon of water (without sugar or artificial sugar) / day. My serum potassium levels remain fine (see below), so instead of going on Potassium Citrate, I’m increasing my lemon juice intake to one cup / day mixed with the same gallon of water, and will do another 24-hour test in a few months.
But, I’m wondering if you think the lemon juice is only increasing my urinary pH, and not helping my citrate, and thereby I should try Potassium Citrate instead? TIA for taking the time and for all your amazing articles! Ps. Potentially I’m one of the unlucky ones in which trying to increase my dietary citrate mostly only increases my pH instead?
August 2021 Results ||| January 2022 Results
Urine Volume: 3.65 ||| 3.99
SS CaOx: 3.73 ||| 1.84
Urine Calcium: 290 ||| 181
Urine Oxalate: 31 ||| 26
Urine Citrate: 579 ||| 259
SS CaP: 1.74 ||| 1.07
24 hour Urine pH: 6.962 ||| 6.890
SS Uric Acid: 0.04 ||| 0.04
Urine Uric Acid: 0.456 ||| 0.494
Na 24: <36 ||| 51
K 24: 103 ||| 91
Mg 24: 129 ||| 153
P 24: 0.847 ||| 1.033
Nh4 24: 23 ||| 45
Ci 24: <55 ||| 68
Sul 24: 39 ||| 36
UUN 24: 9.01 ||| 9.72
PCR: 1.1 ||| 1.2
Normalized Values:
Cr 24: 1420 ||| 1272
Cr 24/Kg: 22.8 ||| 20.5
Ca 24/Kg: 4.7 ||| 2.9
Ca 24/Cr 24: 204 ||| 143
Metabolic blood panel results:
Sodium: 139 ||| 138
Potassium: 3.9 ||| 3.9
Calcium: 9.0 ||| 9.3
Creatinine: 0.95 ||| 0.88
Bun/Creatinine: 14.7 ||| 17.0
Hi Bret, The high urine ammonia and fall in citrate probably reflect potassium depletion. Most potassium is in cells, so serum levels are of no value until cell stores become rather low. I would speak with my physician about using potassium chloride to replace the potassium loss from the diuretic without adding alkali and I would dump the lemon juice – it is an unproven and probably ineffective item. Regards, Fred Coe
So Potassium Chloride, not Potassium Citrate, because Potassium Chloride shouldn’t increase my pH, but still should help increase my urinary Citrate level, because of the Potassium? And also, ceasing the lemon juice should lower my pH a bit too, right?
It will be weird drinking the gallon of water I drink / day without lemon juice in it! But if I’m following what you’re saying, it makes sense. Thanks!
Oh, also, what dosage of potassium chloride do you suggest?
Hi
Of course your physician needs to decide that. A common dose is 10 to 20 MEq twice a day but each person is individual for dosing. Regards, Fred Coe
I have been adding a tablespoon of potassium citrate to my daily smoothie for a year.
My serum potassium is 4.5 and serum chloride is 101.
My urine PH is above range at 8.5.
Is this an issue?
Would potassium bicarbonate have the same result?
Hi Bobby, why do you do this?? The urine pH is very high in part from the K citrate in part smoothies to this and can promote kidney stones and obesity. As I have no information I cannot take things further. Regards, Fred Coe
Hi Dr. Coe. I was wondering where Urinary Citrate level fits in priority for an idiopathic hypercalciuria calcium-oxalate sufferer, as compared to sodium, calcium and oxalate intake? Thanks as always!
Hi Brett, Because calcium binding and an inhibitor of crystallization urine citrate is protective against calcium stones. If your 24 hour urine citrate is below 400 mg/d risk increases, so citrate supplement is worthwhile. Regards, Fred Coe
Dr. Coe –
I’m trying to make sense of some of the new over-the-counter ‘citrate’ products that are being sold, and I can’t really figure out whether these supplements will actually do what they claim. There’s one product (I won’t use the name!) which is a powder with each dose containing 200 mg of potassium ( as citrate), 30 mg of magnesium (as citrate) and 60 milligrams of sodium bicarbonate. They claim that each packet provides ’10 mEq’ of citrate. Can that be true?
Hi Colin, I had begun an article on Moonstone and the like but did not finish it – other things to do. You will find that none provide more than about 3 mEq of potassium per dose because of federal guidelines sans prescription. They work around that with sodium or other based alkali. I have not reviewed what evidence the makers have published about these products, so I am not able to say. It is like this: I have to pick what matters, and so far this has not reached high enough to displace other things. Very sorry as I would have wished to review the OTC alkali by now. Fred
Dr. Coe, thank you for such an informative article. I appreciate your analogies, attention to detail, and sense of humor. Your selflessness in this continuing endeavor is unmatched and a rarity in this day and age. Thank you for spreading knowledge and kindness.
Thank you so much, Mari, and I am glad the site has been a help for you. I do it because the culture supports people like me – professors – by NIH grants and by tax subsidies of universities. I feel one should pay back after a lifetime of such support. That simple. Regards, Fred
Hi Dr.,
I am 45 years old and a pretty fit individual who trains extensively – mountain biking, weight training, rock climbing etc. Recently I have been diagnosed with a 5mm stone in my ureter and 1 additional stone <4mm lodged in my kidney. The pain has subsided and im having 1 gallon of water every day…with lemon | apple cider vinegar. The goal is to try and dissolve the stone so that it passes. I have tried to read and re-read your article a few times, but maybe im not fully grasping it. I have significantly reduced my protein intake. Will supplementing with Potassium Citrate pills help in dissolving the stone ?
Thanks and regards,
Sid
Hi Sid, If uric acid, raising urine pH can help dissolve the stones and will surely prevent more. But if calcium oxalate or calcium phosphate alkali will not dissolve the stones. Your physician can tell if the stone is uric acid by analyzing it after passage or removal or by measuring its density on the CT – every physician can do that. UA stones have low radiographic density. Regards, Fred Coe