

I do not know if Edvard Munch (1863 – 1944) had Lot’s wife in mind, but I do and thought this an apt image.
She crystallized, perhaps into calcium carbonate, hence my putting her here at the beginning of our sojourn into the vast territories of calcium metabolism and the hypercalciuric states. If I were to single out one condition that dominates stone disease prevention because of sheer commonness and reliable means of treatment it would be this one.
I have put off the crucial topic of hypercalciuria for the first year of this site because I wanted to build a proper foundation.
We now have good materials about stones and stone crystals, supersaturation, and certain critical treatments such as potassium citrate and high fluid intake.
It is time to tackle the problem of high urine calcium itself, what happens when it is high, why it might be high, what levels pose stone risk, and how it is best treated so far as we know.
This article lays out the relationships between urine calcium and stone risk, names the important causes of high urine calcium in stone forming people, and the other problems high urine calcium may be related to.
It is an introduction: Long, but brief in comparison to what is a major component of stone disease and its clinical management.
What is Hypercalciuria?
Stone Risk
To me the most immediate definition is about stones: At what level of urine calcium is the risk of new stone onset increased?
The best information to date arises from long term longitudinal data contributed by two cohorts of nurses and one of physicians exploited by Dr. Gary Curhan. In each cohort, some people became stone formers, and most did not. Gary obtained 24 hour urines in a cross section from all three cohorts, and was able to relate the level of urine calcium excretion to the risk of forming stones.
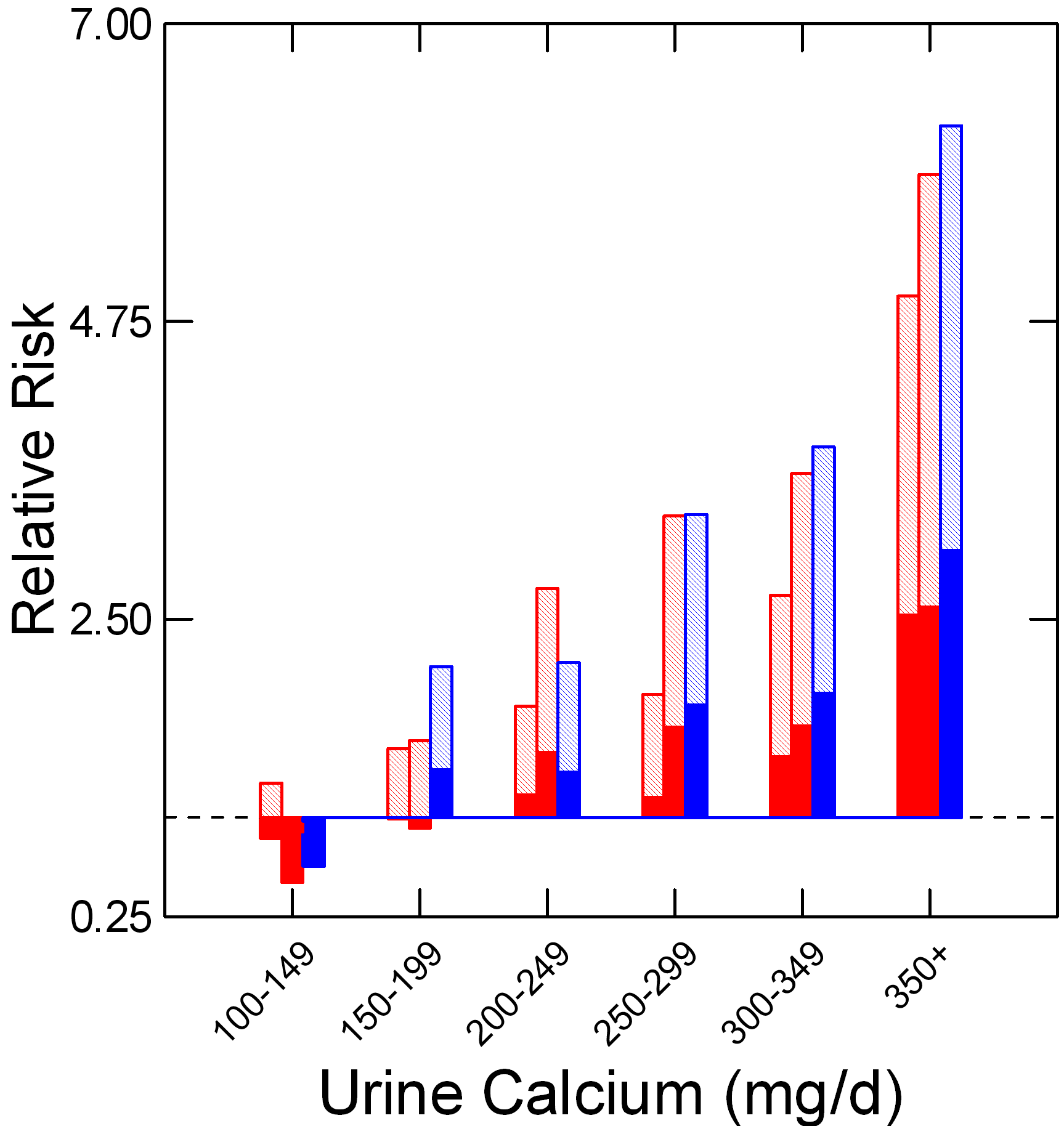 Along the horizontal axis urine calcium excretions are groups into 6 bins; above each bin the relative risk of becoming a stone former is presented for the two nurse cohorts (all women, and in red bars) and the physician cohort (all men and in blue bars). The dashed line is at 1, meaning the baseline of risk for the population which was at taken at <100 mg/day of urine calcium loss.
Along the horizontal axis urine calcium excretions are groups into 6 bins; above each bin the relative risk of becoming a stone former is presented for the two nurse cohorts (all women, and in red bars) and the physician cohort (all men and in blue bars). The dashed line is at 1, meaning the baseline of risk for the population which was at taken at <100 mg/day of urine calcium loss.
The relative risks are given in the manuscript as an average (mean) value and an upper and lower 95% confidence limit. But I am showing only the lower 95% confidence limit (solid bars) and means (lighter bars).
For example, in the 100-149 bin the mean relative risk for the first of the two female (red) cohorts is 1.26 (plotted up from 1 as a light bar), the lower limit is 0.84 (plotted down from 1 in a solid red bar) and the upper limit is 1.91 (not plotted).
I am after a measure of risk that is robust: In which bin is it very likely that risk is increased above the 100 mg/day baseline?
For me, that is when the lower limit for the relative risk is above 1.
For example, the value of the lower 95% risk confidence limit of the 100-149 bin, in the first of the two nurse cohorts – 0.84 – does not connote a high likelihood of significant risk compared to 100 mg/day of urine calcium.
On the other hand, risk is very likely present above 200 mg/day of urine calcium, because the lower 95% limit is above 1 in all three cohorts. Of interest, women and men do not seem to differ, so this one criterion applies to both sexes equally.
Also of interest and great importance, there is a dose response.
Higher and higher urine calcium levels are associated with higher and higher relative risk. This is good supportive evidence of a causal connection and best seen by the position of the means which are at the tops of the bars..
The direction of causality is not determinable from observation, but I vote for higher urine calcium causing stones not that being a stone former somehow raised urine calcium.
My vote is because of the other science.
This site has belabored the point that supersaturation drives crystallization, and stones are made of crystals. In preparation for this new series of articles on urine calcium I have gathered the main articles on the stones themselves and on supersaturation into a pair of ‘walking tours’ which present them in context and with with commentary. These are the foundational articles upon which the effects of urine calcium and other urine constituents can be assembled to produce a reliable picture of how stones form and how they can be prevented.
Here, I simply note that higher urine calcium loss will increase average urine calcium concentration for any given urine flow rate, and higher calcium concentrations will in general produce higher SS values for calcium phosphate and calcium oxalate and therefore higher risk of stones. So hypercalciuria, to me, and for good reasons, is almost certainly a cause of calcium stones via increase of SS which is the prime measure of the energy that drives crystallization.
Upper End of the Normal Range
A valid and alternative definition of hypercalciuria is that it consists in very high urine calcium excretion, and a way to gauge the meaning of ‘very high’ would be values at the upper end of the normal range. This idea of the ‘upper end’ is usually taken as above the upper 95% of values encountered in surveys of people without known diseases.
The Curhan data actually give a reasonable measure of this ‘upper end’ from the means and 95% limits of the non stone formers in the three cohorts. The lower dot marks the position of the lower 95th percentile of 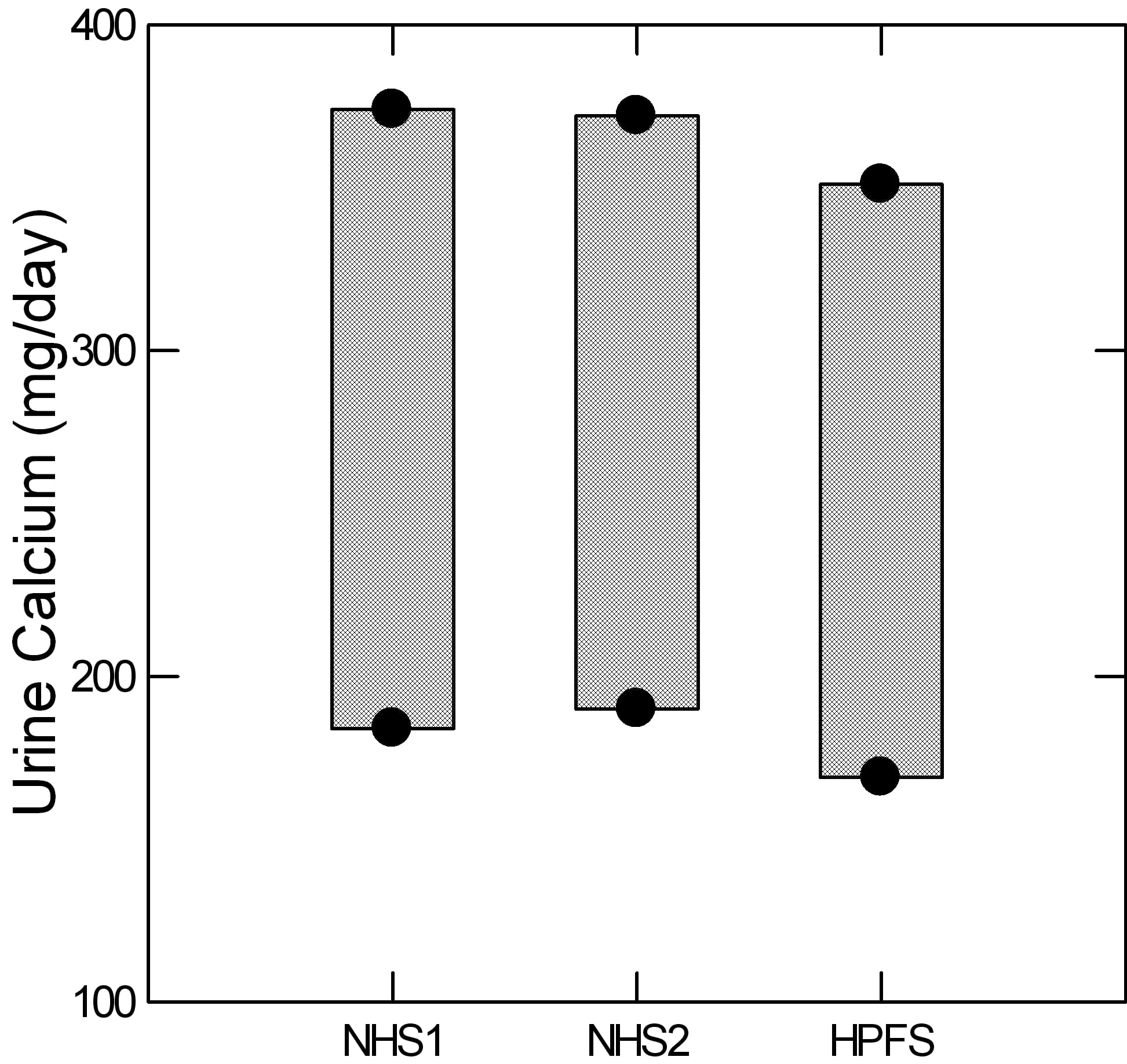 urine calcium for the two nurse cohorts and the physician cohort. The upper dot gives the upper 95th percentile (two standard deviations above the mean for those technically inclined). The intervening bars are for visual effect.
urine calcium for the two nurse cohorts and the physician cohort. The upper dot gives the upper 95th percentile (two standard deviations above the mean for those technically inclined). The intervening bars are for visual effect.
For those who want to study hypercalciuria, as an example, and want a reliable gauge of who is ‘really high’ the three figures are 374 372 and 351 mg/day for the three groups.
Older Conventions
Using smaller and less well selected populations, many investigators have derived somewhat different upper limits for defining hypercalciuria. For example urine calcium excretions above 250 mg/day or above 4 mg/kg body weight are common as in this typical kind of research publication. In our own reviews we have frequently used 250 mg/day for women, 300 mg/day in men, and 4 mg/kg body weight either sex as upper limits to define hypercalciuria.
Overview
I think that the Curhan values are ideal for clinical practice. People with calcium stones and urine calcium levels above 200 mg/day have increased risk from their urine calcium and one aspect of their treatment can be to reduce urine calcium.
That is to say, physicians look at all possible factors that may be increasing risk in a patient, sort them out and select for treatment those most promising and practical to treat. Among them will often be urine calcium and Curhan has given us good targets and criteria.
For research, I am more flexible. Strictly speaking the 95% upper limits are a kind of ideal, but if you want to understand mechanisms that raise urine calcium one might be better served by selecting cohorts with a range from high normal to very high. These are research design decisions, and all I mean to say here is we have decent ideas about normal ranges and stone risk, and it is possible from them to derive appropriate approaches to clinical research on hypercalciuria.
Diseases That Cause Hypercalciuria in Stone Formers
Let me be clear about goals here. Each of these diseases will get its own articles, or perhaps many articles, so this is not meant to be a proper exposition but merely an introduction.
For this reason referencing is light and I mainly make assertions which are commonplace and can be found in any textbook or review article. When I get to the diseases one by one I hope to offer considerably more than is commonplace.
Primary hyperparathyroidism (PHPT)
About 3 – 5% of calcium stone formers have this curable disease, on average, so its detection is crucial for proper patient care. As a reference I have used my own publication because it contrasts patients with this disease to ordinary stone formers, is available as a free pdf, and, perhaps, because I wrote it.
One or more of the parathyroid glands enlarges and produces an excess of parathyroid hormone (PTH). PTH signals kidney cells to retain calcium by reabsorbing a higher fraction of calcium that is filtered by the glomeruli. It stimulates production of the active form of vitamin D (calcitriol) and that in turn increases the efficiency of GI absorption of calcium from foods. Finally PTH increases bone turnover so more calcium than normal leaves the bone and can be lost in the urine.
The increased bone calcium loss and increased GI calcium absorption are balanced by increased urine calcium losses, and these can be very impressive. The high urine calcium losses raise urine supersaturation with respect to calcium oxalate and calcium phosphate so stone risk rises and patients with this disease not uncommonly present themselves as stone formers.
High rates of mineral loss from bone cause bone mineral depletion, and bone disease is well known to occur. If the disease is cured in a timely way bone healing is expected.
The action of PTH to increase the fraction of filtered calcium that is reabsorbed causes blood calcium to rise. The increase is often modest.
Being rarely malignant, the enlarged parathyroid glands can be removed surgically with expectation of a cure, so this disease is a curable cause of kidney stones and bone disease.
The diagnosis depends upon finding high urine calcium excretion, a serum calcium concentration above normal for the laboratory making the measurement, and a serum PTH value which is not suppressed below the normal range.
However, there are a few cautions that must be considered always. Even if everything I have just said is true, there are artifacts that lead to mistakes in diagnosis and could lead to unnecessary surgery. All thiazide type diuretics can raise serum calcium, so testing needs to be done after 2 weeks off the drug. Lithium can raise both serum calcium, serum PTH and even urine calcium, so is a real fooler. Hyperthyroidism, including that induced by too much thyroid hormone replacement, can raise serum and urine calcium although it does suppress serum PTH. The serum for PTH must be the one for the calcium – same blood draw for both, and must be drawn fasting overnight.
The relationship between PTH and serum calcium is often misunderstood. The calcium balance systems are elaborate and tend to be self regulating, so increased PTH secretion leads to increases in serum calcium and calcitriol both of which, through negative feedback on the glands, bring PTH down toward or even to normal, but never below normal. Therefore PHPT is diagnosed when the serum calcium is above normal and the PTH is not below normal.
Here are the serum and urine calcium values from the PHPT cases in our publication – all proven surgically – shown in large dots, normal people (medium dots) and thousands of points from stone formers without any systemic disease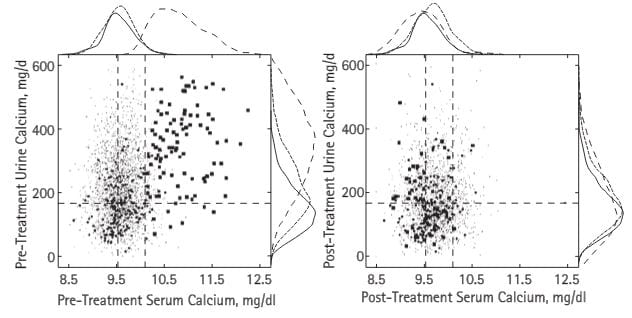 – fine spray of tiny dots (common stone formers). The vertical dashed lines are the normal range for blood calcium in our kidney stone laboratory, the horizontal line is just below the level where stone risk from urine calcium begins (200 mg/day).
– fine spray of tiny dots (common stone formers). The vertical dashed lines are the normal range for blood calcium in our kidney stone laboratory, the horizontal line is just below the level where stone risk from urine calcium begins (200 mg/day).
The patients all had serum calcium values above normal but many were just slightly high. Urine calcium was a lot higher than in ordinary stone formers, but even in those the plume of fine dots rose far above 200 mg/day. Note that some normal people had values above 200 mg/day.
After surgical cure (right panel) the PHPT cases, normals, and treated common stone formers who had no systemic disease all contracted into a small region, but not a few urine calcium values were above the fateful 200 mg/day limit still.
The curves outside the boxes show the distributions; see how the hyperparathyroidism points (dashed lines) spread out far above those for normals and ordinary stone formers, then fall back with surgical cure.
Normocalcemic Primary Hyperparathyroidism
Properly speaking this would be high urine calcium excretion, normal blood calcium, and reasons to think that an abnormality of parathyroid gland function was responsible for the high urine calcium – which is the only abnormality. One such reason could be an elevated level of serum PTH, and such levels are encountered from time to time. Another could be blurring of the true upper limit for serum calcium; not all laboratories are equally crisp. A third could be, and this is speculative, that along the course of PHPT glands might lose their normal CaSR responsiveness, so that PTH levels are no longer down regulated normally by serum calcium. In another article I will explore this problem. Right now, I believe it is wiser to wait for hypercalcemia before committing to neck surgery in hopes of cure and treat the hypercalciuria as if it were idiopathic hypercalciuria – see below. PHPT will ultimately declare itself in most cases. The reference in the link above is supportive of my suggestions because the authors expect elevated serum calcium in PHPT, as I do.
Secondary Hyperparathyroidism
Being so caught up in the maintenance of a normal serum calcium, the parathyroid glands will react to any threat with increased or decreased PTH secretion as required to restore that normal value. Low calcium diet, vitamin D deficiency, GI diseases with calcium malabsorption for any reason, chronic kidney disease with its odd form of calcium aberrations, all these will and do cause increase of PTH secretion and raised serum PTH levels. None of these are hypercalcemic states, and most have a low urine calcium loss. Stones may be present but for reasons other than parathyroid abnormalities, and often other than level of urine calcium. For example, acid urine pH from kidney disease can cause uric acid stones; high urine oxalate from bowel disease can cause calcium oxalate stones. All I mean to say here is that isolated increase of serum PTH with low or even normal urine calcium and normal or low serum calcium levels are usually not an occasion for parathyroidectomy, but call for a diagnosis of their own. As I have by said to excess, this is an issue that deserves more than an introduction. The link for normocalcemic primary hyperparathyroidism is useful for this topic and the one below.
Familial Hypocalciuric Hypercalcemia (FHH)
High serum calcium with low urine calcium excretion – below 100 mg/day is common – is almost never primary hyperparathyroidism but rather a mutation of the PT gland CaSR that raises its sensitivity so serum calcium can be low, serum PTH normal, and urine calcium quite low. When FHH patients have stones the stones are not due to high urine calcium but some other cause, and most importantly parathyroid surgery is a mistake.
Sarcoidosis
In my long experience I have seen far less than 1% of calcium stone formers present with this condition. A reputable source of information about Sarcoidosis fails to mention kidney stones so far as I could see.
Briefly, sarcoidosis is a disorder of the immune system and the cells which proliferate and enlarge lymph nodes, liver, spleen and other tissues produce calcitriol. So the physiology is that of unbalanced high calcitriol production.
As I have mentioned above, calcitriol increases GI calcium absorption, so food calcium entry into the blood increases. I have not mentioned but say now that calcitriol increases net bone mineral losses mainly by reducing bone production. Calcitriol is a steroid hormone and like many steroids acts on the nuclei of cells. It acts on parathyroid cells to shut down production of PTH so tubule reabsorption of filtered calcium is reduced as compared to primary hyperparathyroidism.
The increases in bone mineral loss and GI calcium absorption raise urine calcium excretion as in hyperparathyroidism. Because PTH is suppressed below normal, serum calcium may not rise or when it does so rises only slightly at first.
This means that in many instances suppressed serum PTH may be the only laboratory clue to sarcoidosis as a cause of hypercalciuria and stones. Clinically sarcoidosis is often diagnosed from its signs and symptoms, as noted in the reference from the National Heart and Lung institute. I have had cases which I diagnosed de novo being without obvious signs otherwise because of suppressed PTH with very high urine calcium and even elevated serum calcium.
Serum calcium rises when the flows of calcium from bone and GI absorption are very marked or when the efficiency of renal calcium removal falls.
This latter can occur because of calcium itself. It can reduce water and salt conservation and therefore reduce the blood volume and therefore filtration. Any reduction will tend to increase serum calcium, and increases in serum calcium will reduce filtration and water losses, so a kind of unhappy cycle begins. How calcium acts to reduce calcium reabsorption and that it also acts on filtration itself comes later, not in this article but in those perhaps months from now.
CYP24A1 Deficiency
Vitamin D3 is made in the skin or consumed in pills, and in the liver converted into 25 hydroxy (OH) vitamin D (25(OH)D3). The enzymes responsible for the conversion are called CYP2R1 and CYP27A1.
25(OH)D3 circulates and is itself biologically active, but has a larger fate. Some is converted by kidney cells into 1,25(OH)2D3 (calcitriol for ease of writing and reading) and it is this molecule which more powerfully activates tissue responses in the GI tract, kidneys, bone, and – as already mentioned – parathyroid cells. The enzyme which activates 25(OH)D3 to calcitriol is called CYP27B1.
CYP24A1 is a general purpose inactivator of 25(OH)D3 and calcitriol. It converts the former into inactive molecules in liver. In kidney it converts calcitriol into other inactive molecules. If this degrading enzyme is deficient because of mutations, calcitriol levels increase as in Sarcoidosis, and one gets a similar picture of high urine calcium suppressed serum PTH and high normal to high serum calcium, with kidney stone formation as a consequence of the high urine calcium.
This is a rare condition; in my lifetime of practice I have encountered only 2 cases I know of.
Vitamin D and Calcium Supplement Excess
Certainly, these OTC materials are associated with increases of urine and even serum calcium, especially noted in menopause when their use is prevalent. Causality is not certain. For example, serum 25(OH)2D3 levels between 20 (a low number) and 100 (a high number) were not associated with kidney stones in a small sample of 2012 people. However in a large trial, it appeared that calcium supplements, not food calcium, might be specifically a risk for stones. Calcium supplements might be a particular hazard in people prone to high urine calcium excretion by their genetics.
Distal Renal Tubular Acidosis
I have referenced our paper not out of community pride but because it is the only one which describes the papilla and separates stones from nephrocalcinosis. In a massive surgical practice at the Kidney Stone Center at Indiana University we could find only 5 cases on whom we have operated and obtained research information about the papillary tissues.
All five had the expected reduction of serum bicarbonate with increased serum chloride (no elevated anion gap), alkaline urine pH, and reduced serum potassium – from the obligatory bicarbonate driven kaluresis. Several were hypercalciuric.
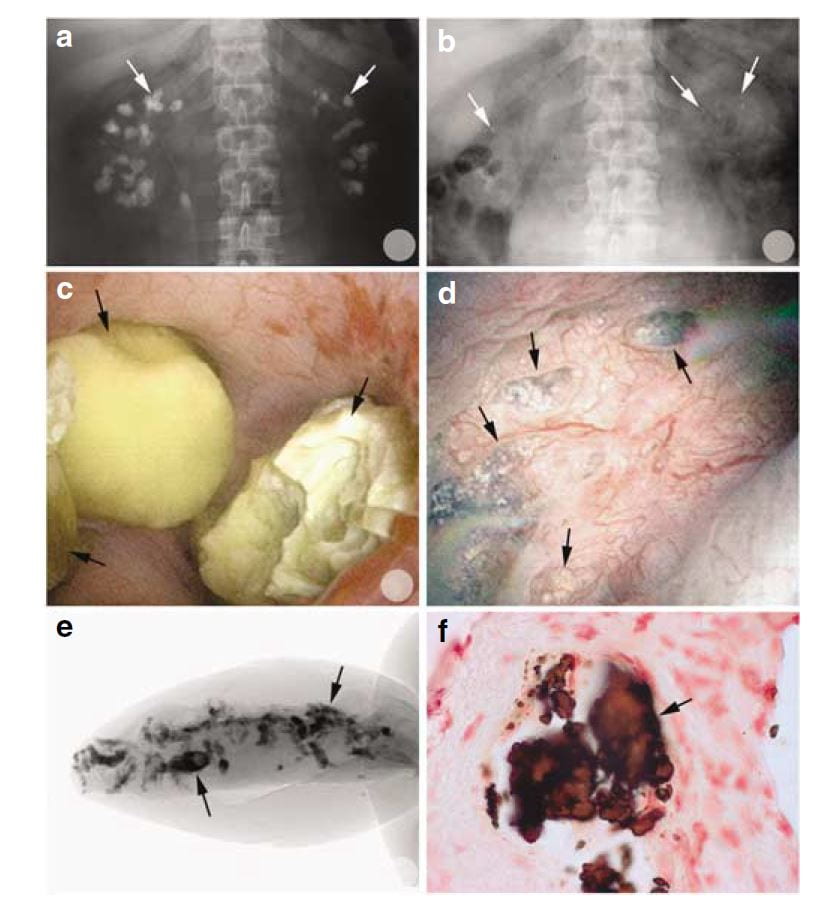 Of interest, most of the calcifications seen on radiographs were, at surgery, removable stones as opposed to crystals embedded in the renal tissues.
Of interest, most of the calcifications seen on radiographs were, at surgery, removable stones as opposed to crystals embedded in the renal tissues.
You can tell this from the x rays in the upper two panels. Before percutaneous nephrolithotomy nephrocalcinosis was dramatic; afterwards, few calcifications were present.
In the middle left panel you can see stones in the calyces during surgery, and in the right middle panel you can see the papilla after the stones were taken out. It has some calcifications in the tissue which are plugs in tubules. These plugs are shown by a micro – CT of a tiny biopsy from the papilla (lower left) and in a histological section (lower right)
Stone cultures were positive, perhaps because these patients had many prior procedures for multiple stones.
Deposits of calcium phosphate crystals affected virtually all of the Bellini ducts and though each deposit was small plugging on average replaced much of the papillary tissue. Despite this, kidney function as measured clinically was not remarkably abnormal, and the cortex of the kidneys not remarkably damaged.
In 1980 I presented our only 6 patients with stones and distal RTA out of over 1,000 stone formers to date, of whom 4 were from one family. They all had low bicarbonate and high chloride in their serum, as expected, an alkaline urine pH, and were unable to lower the urine pH with an acid load. Only one was convincingly hypercalciuric.
The essence of dRTA in stone formation is this: By inheritance or because of Sjogren’s syndrome, SLE or other immune mediated diseases the ability of the collecting ducts to lower urine pH normally is diffusely impaired, not in the patchy way one might expect from tubule plugging with crystals but throughout the medulla and papillae. High tubule fluid pH and, when present, hypercalciuria raise supersaturation with respect to calcium phosphate and diffuse plugging occurs with considerable damage and loss of tissue.
Perhaps because of its diffuse nature, dRTA does appear to lead to overall reduction of renal function if enough cases can be found and comparisons made to other forms of stone disease. In this study we had 1,856 patients with stone analyses and renal function, as well 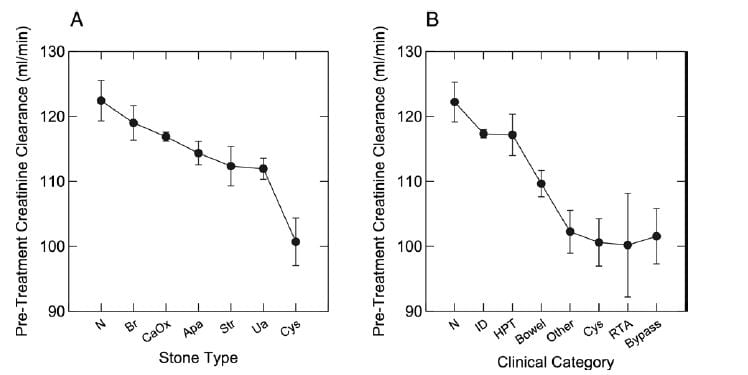 as diagnoses, all from our Kidney Stone Prevention Program at University of Chicago – the program that brings you this website.
as diagnoses, all from our Kidney Stone Prevention Program at University of Chicago – the program that brings you this website.
Measured creatinine clearances are shown as means with standard errors for all stone types on the left, and by diagnostic category on the right. Note that RTA, cystinuria, obesity bypass patients, and a miscellany of other rare disorders (like sarcoidosis and CYP 24A1 defects) shared a distinct reduction of renal function, whereas the common calcium stone types without systemic diseases, and even primary hyperparathyroidism (HPT), had no reduction compared to normal (N). ID refers to idiopathic hypercalciuria, people who have high urine calcium losses without obvious disease.
What does all this mean?
Distal RTA is very uncommon, colorful, easily diagnosed by blood abnormalities, family history of like diseases, and the presence of immune diseases. The label RTA may be misapplied to patients with many calcium phosphate stones because they produce an alkaline urine pH; that is not true RTA, but how phosphate stone formers are.
More Rare Genetic Mutations
These conditions are almost always evident in childhood, very rare, and not what a clinician in the practice of stone prevention expects to encounter. However every once in a while they do show up. The best reading source for all of the genetic hypercalciurias is OMIM, the wonderful online library that is free to everyone. In reading the tiny blurbs below keep in mind that almost no stone formers have these rare diseases, that those who have them present typically as ill children, even infants.
Bartter Syndromes 1-3
These are gene defects of transporters in the thick ascending limbs of the loops of Henle (In case you forgot or never knew the nephron segments this article has a nice picture). There is hypercalciuria but also a picture that resembles lasix use because lasix acts on this segment. Urine losses are high for sodium, potassium, and chloride, and people with these diseases are prone to low blood pressure if they do not get enough sodium replacement. The blood bicarbonate is high, potassium is low.
Bartter Syndrome 5.
There is excessive signalling of the cell surface calcium receptor (CaSR) which produces a lasix like picture but because of the specific problem serum calcium is low and so is serum magnesium. The parathyroid glands are regulated by serum calcium via via the CaSR which is the reason for the low serum calcium level.
Autosomal dominant hypercalciuric hypocalcemia
The CaSR is altered genetically but the specific alteration causes a more marked change in serum calcium than in thick ascending limb function, so the serum potassium and bicarbonate can be normal. These two diseases BS 5 and ADHH are essentially on a spectrum and resemble each other.
Dent Disease
A genetic mutation in a chloride transporter leads to hypercalciuria and, in males especially, progressive kidney disease. The serum values are normal apart from reduced kidney function.
Medullary Sponge Kidney
Given the nephrocalcinosis and many stones, one might think MSK is a cause of extreme hypercalciuria, but that is not the case. Among our 12 cases that were proven to be MSK during surgery and with papillary biopsy, the average urine calcium was 250 mg/day, just above the Curhan threshold for significant risk. SS values for calcium oxalate averaged 7 and for CaP 1.3, values scarcely above those encountered within normal populations or among common uncomplicated calcium stone formers.
We noticed the same in our much earlier paper on MSK diagnosed in the era of intravenous pyelography. We have already mentioned that among our two MSK series serum levels showed none of the abnormalities of renal tubular acidosis (low bicarbonate and potassium) and urine pH was not high (6.1 in our recent series). Ductal and clinical stones were on average calcium oxalate, not calcium phosphate. We believe the stones form in the stagnant recesses of the collecting duct cysts, abetted by a very modest bias of urine calcium excretion above the normal median value. This is what one expects when supersaturated urine is trapped in place, and supersaturation inevitably expresses itself in gradual crystallization.
Idiopathic Hypercalciuria (IH)
What is It?
In those two words you can find the major preoccupation of my life as a clinical investigator. To the mechanisms responsible for this common abnormality I have devoted my energies since I first began, in 1969.
The reference in the link above is to a review by my colleague Dr. Elaine Worcester whose brilliant research has clarified how the kidney goes about raising the urine calcium in this condition.
IH is not a disease.
It is a stone risk.
And that risk can be ascertained in any given person from the Curhan data in the first figure in this article.
People with high urine calcium excretions who do not have any of the diseases I have mentioned, or any of the others I have not – for pity and reasonable length of this article – mentioned thus far, have hypercalciuria that is ‘idiopathic’, of or from themselves or itself if you wish. They are the top of the mark among normals.
No red or green line separates IH from normal. Just as high blood pressure is a risk factor for stroke and heart disease, hypercalciuria is risk factor for disease – stones and bone disease, which is why I have devoted to its study so much of my life.
Being one end of the human distribution, IH may be found in your next door neighbor, or even your spouse and you would not know unless they are tested. And they will not be tested unless they get stones, or bone disease, or their family members get these diseases. If testing is done nothing will be found but high urine calcium, enigmatic, distinctive yet bland, but bespeaking a complex physiology.
Where Does the Extra Calcium Come From?
In the named diseases, hypercalciuria comes from bone and food calcium, and the same is true for IH. It has to be so, there are no other sources. But IH is not a disease, so we are seeing in IH a magnification of the usual losses of bone and diet calcium in urine, meaning that one or both of these must be increased. Certainly, as I will show in other articles, diet calcium is absorbed more efficiently in IH than in normal people. But bone mineral can be lost in excess, and bone disease can result.
It is for this reason, Dr. David Bushinsky, a distinguished bone and mineral investigator and internationally recognized authority in calcium metabolism, has proposed that every stone clinic is a bone clinic. Stone formers have the potential to develop bone disease if not properly cared for. Though their stones are an immediate reason for attention and a prime focus of care, bone health is also an inescapable concern.
How Can The Urine Calcium Be Lowered?
Toward the lowering of urine calcium and consequent prevention of stones – and preservation of bone mineral – we have some useful trials using thiazide diuretic type drugs. Likewise, some trial evidence favors reduction of diet sodium intake, to which I have alluded without presenting evidence.
Why Are We Ending Here?
It could seem ungracious to end here, without more details about IH or the many named hypercalciuric diseases, but this article is already very long, and what remains is a massive amount of information. I plan for many articles on hypercalciuria, the named diseases that cause it, and especially on IH itself because of its central role in stone disease and because it is a personal interest of mine and of those I work with.
Here I mean merely introductions, as at a cocktail party.
It seems probable to me that in doing right by this topic the articles on this site could double in number, although that may be an overestimate.
Check by from time to time, and you will find more.
Regards, Fred Coe
I am a stone former and I have had 3 stones since 2017. All passed without surgery but I was not able to have them tested. They must have broken up. Anyway, before I heard of oxalate I ate a lot of high oxalate foods. I’m now on the low ox diet and eat more calcium. I recently had my vitamin D level tested and it was a bit low at 25 ng/mL. I was told by my PCP to supplement with 2000 IU of D daily. I’m reading a lot of contradictory information online about whether vitamin D supplements increase stone risk. Can you please discuss this? Thanks in advance!!
Hi Morris, Apart from overt excess, vitamin D supplements have not been associated with increased stone risk. Levels of 35 – 40 in the serum are almost certainly not a concern. Regards, Fred Coe
Hi, I am a 37 yr old female and I have kidney stones. My parathyroid levels are normal. My vitamin d is 27- a little on the low side. I had a 24 hour urine analysis and my calcium level was 322, pH 6.9 and SS CaP 1.72. Everything else was normal. I had my genetics done through 23andme and also through a study at u of m. I put the results through a website called promethease – that collects all studies done on genetics. Both results show that I have Hyperglycinuria (or more rarely, iminoglycinuria) – a tendency towards kidney stones. My question is do you know anything about this? Is this why I produce stones? What can I do? Thank you!
Hi Natalie, OMIM details the gene defects in SLC36a2 but in the papers I found no strong link to stones. This excellent JCI paper examines the relationships between multiple gene disorders and the urine loss of proline and glycine, but nowhere mentions kidney stones. I suspect your stones arise from your marked hypercalciuria and alkaline urine and would suggest your physicians focus on treatments for those conditions. Regards, Fred Coe
I really appreciate the reply. The nephrologist I see told me D supplements can increase urine calcium and should be avoided.
Dr. Coe,
I really appreciate your informative articles. I had two kidney stones in 2017 at age 48. I passed one and the other is a 4mm non symptomatic stone in the lower pole of my left kidney that has been stable for almost 2 years. For the past many years, my blood calcium has been rock solid between 9.1 and 9.6 without much variance and my PTH has been 32-54 range, ruling out hyperparathyroidism. However, I have done eight 24-hour urinalysis tests over the past 2 years following the stone onset with urine calcium typically in the 260-320 range with one outlier measurement of 197 and one at 616, and oxalate levels in the high 40’s. Citrate excretion is in the 500’s and PH is just under 6. Urine volume has always been over 3 liter, so concentrations are always low and supersaturation analysis comes back “normal”, with no increase risk of any stone formation. I do not take calcium supplements, but eat 1000-1200mg of calcium in food a day (yogurt, cheese, whey protein (keep protein intake to less than 100 grams a day). I am now 50, but extremely athletic (I bike 150 miles a week and lift weights and am 6’5″ 200 lbs). I also keep sodium to less than 2000mg a day and oxalate is between 50-100mg a day. I am still concerned about future bone density loss with the 300 ish mg of urinary calcium daily. I would like to stay away from thiazide diuretics as the side effects for men are significant. Is osteoporosis a certainty at this level, and is there anything else I can do to prevent bone loss? Because of my good PH levels, and high daily urine output, I feel like I am diluting the oxalate and calcium enough to keep stones away, but I want to stay active and not risk breaking bones in the future. Would you recommend a bone density scan or any other course of action at this point?
Hi Steve, You appear to have genetic hypercalciuria. In this condition bone mineral loss is not rare, and a routine DEXA scan a good idea. For stone prevention, you are doing low sodium high diet calcium but be sure your 24 hour urine sodium is really 2000 mg. Regards, Fred Coe
Thanks for the detailed information. I have been through a mass of the articles relating to stones. I have had two or three stones pass – calcium oxalate – (35yr Male). I have been told I have either two 5mm stones or one 9mm in one kidney and 2x 4mm in the other. My Nephrologist put me on Chlorthalidone to try and reduce my urine calcium levels which were on the upper end of normal. I believe the rating was 7.5, but forget the scale or measurement. Six months in and the drug hasn’t dropped that level, but has caused me to lose a significant level of potassium for which I supplement. My doctor is at a loss as to any other help he can give me as he feels raising the dosage of the drug will just cause more potassium loss. My urine oxalate levels are so low they’re off the scale. “the lowest I have seen”, as my doctor said. The only other remarkable item that consistently shows up in my tests is low platelet levels in my blood. I avoid oxalate items, no cola’s, I drink 3L of lemon juice/coconut water daily, exercise regularly/vigorously. Any advice you could offer to me for these values? Thanks in advance!
Hi Dave, The usual reason for drug failure is too much diet sodium. You presumably have genetic hypercalciuria, and your urine calcium varies with your urine sodium = diet sodium – with a higher than normal slope. Urine sodium often needs to be at the US ideal level of 1500 mg (65 mEq)/d. Higher causes potassium wasting and failure. This is why I advocate for the kidney stone diet first, and drugs as needed thereafter. Lemon juice sounds like a bad idea because of the high sugar load, unless you know otherwise about the product. Sugar raises urine calcium. And, why? No data and an awful lot of lemons! The low platelet count if significant quantitatively needs attention as to cause, and usually hematologists do this. Take a look at the links and see if this works for you. Regards, Fred Coe
Hi Fredric,
Thanks for the reply! While I try and keep as low sodium as I can, it is likely still too high and perhaps that’s why. It’s interesting that you say that also relates to potassium loss. The lemon juice was substituted instead of potassium citrate pills and the juice has no sugar. I will have a look at the links provided. Thanks again!
Hi Dr. Coe:
I am 61, have been vegan for about the last 2 years and had my first stone (4mm, removed via ureteroscopy and lithotripsy) this past July – 60% cal oxalate dihydrate, 20% calcium phosphate (hydroxy- and carbonate-apatite), 20% ammonium acid urate. My 2 24-hr urine tests showed urine calcium of 453 and 508, with PH readings of 5.991 and 6.738. Blood calcium is stable at 9.9, PTH good at 5.5. Urine sodium was 84 and 100. I have had IBS and Interstitial Cystitis and low IgA, so was checked for Celiac Disease and fat in stool, both negative. I have osteopenia which has spread from spine 10 yrs. ago to include neck recently. Trying to avoid medicine. My question is, how much calcium should I be aiming for in my diet? I had thought 1200 mg/day; but have read that it should be more like 800 to minimize stones, and that excess calcium (presumably over 800) can also create stones. I feel like that’s a big difference daily that I can’t afford to get wrong. Is there any difference in daily calcium amounts for treating hypercalciuria with no stones vs. with stones? Also, who should I be seeing about my osteopenia, given that I now have the additional stone problem: a nephrologist? endocrinologist? Have not had any treatment for this issue and feel like I need to be followed, and that diet is critical. Thank you for any help you can give!
Hi Mary, You no doubt have genetic hypercalciuria, but also you may be in a phase of rapidly progressive bone mineral loss as your urine calcium is very high despite a rather modest urine sodium loss. Your problem is not diet calcium intake, it is larger than that, and I hesitate to be too specific from so distant a vantage point. I would consult an expert in bone disease concerning the value of a bone directed medication to stabilize bone mineral and see if that improves your urine calcium loss. I would try to lower diet sodium more, to bring the urine down to about 55 – 65 mEq (1500 mg)/day and consider chlorthalidone at a low dose of perhaps 12.5 mg. The ammonium acid urate is peculiar and suggests diarrhea, laxative use, potassium loss, or perhaps a poor stone analysis. Diet calcium needs to be high, and diet sodium and meds used to control the urine. All this is generalization, and you need a real expert to actually care for you. Regards, Fred Coe
Dear Dr. Coe:
Thank you for your detailed and caring response; I really appreciate your time. I will follow your advice; just wondering if you could say in what department I would find an “expert in bone disease”? Would this be an endocrinologist, or nephrologist, or someone else? Just not sure where to look. Thank you so much again.
Mary
Hi Mary, Oh my, not so easy a question! Bone people are few – at least good ones. Usually they call themselves endocrinologists, but that is very local, and almost always they work in medical schools as the work is technical. If you say where you live I would try to identify someone. Regards, Fred Coe
I’ve suffered from kidney stones most of my life–first at age 20, nothing until 32 and then age 37 and 43 and now at 46. It seems to be speeding up. It’s only been in the past three years that we’ve managed to catch a stone and have it tested. They seem to be mixed oxalate and phosphate (not sure of the %). Urine tests show elevated calcium with normal ranges of everything else. For the past three years I’ve followed a restricted oxalate diet (not sure I even need this), limited my sodium, increased my H20 and calcium (through dairy). With all this, I’m still producing stones. Very frustrated and at the end of my rope.
Hi Jason,
Have you had a 24 hour urine collection? It may help solve the mystery.
Best, Jill
Yes, it’s been done about 5 times now. Always shows elevated calcium and everything else within normal ranges–including oxalate.
Hi Jason,
The high urine calcium may be one of the problems- talk to your doctor about this and what can he/she do to get it under control.
Best, Jill
Hi Jason, You have presumably idiopathic hypercalciuria if your serum calcium is absolutely normal. It is treated with diet and if that fails with meds. Here is a good article on treatment. Regards, Fred Coe
Hi! I am at a loss for what’s going on with Me? I have passed at least 30 stones since i was 18 (am now 37). My pth levels aren’t at 40 and my blood calcium is 10.1 both are normal i believe. I have done three calcium urine tests and all are around 297? I also have muscles that randomly twitch? Could that be associated with to much calcium in my urine? My urologist said my kidneys leak calcium could my body not be getting enough calcium amd that’s what’s going on with my muscles? They want me to take thiazide. What are your thoughts?
Hi Shannon, You probably have idiopathic hypercalciuria and can control it with diet and – if needed – thiazide. The borderline serum calcium caught my attention, as it may signal primary hyperparathyroidism. Serum PTH can be normal in that condition. I would urge several morning fasting serum calcium levels to be sure your value is below 10, or certainly below 10.1. If it is idiopathic hypercalciuria improves with reduced diet sodium, and there is a risk of bone disease so diet calcium needs be high. Regards, Fred Coe
Hi thanks for your reply, just seeing it now for some reason. I started to talk a calcium pill and the muscle twitching almost completely stoped. Could loss of Calcium cause muscle twitching? I will take some PTH blood tests. Is 10.1 high?
Hi Shannon, the serum calcium you mention is a bit high if fasting in the morning. Muscle twitching is not common with increased serum calcium. Regards, Fred Coe
I am 34 year old man. Suffering from recurrent renal calculi, hypertension and severe osteopenia. I had past 2 surgeries for stone. Since last 1 year my serum calcium varies between 9.5 mg/dl to 12.5 mg/dl and pth between 37 to 97. Urine calcium also high 356 mg/day and 375 mg/day.
Hi Govind, with serum calciums this high you must have some underlying disorder. The most likely is primary hyperparathyroidism. That best fits the normal to high PTH, and normal to high serum calcium. The article makes clear how you need to get fully tested and if it is PHPT one can cure it. Regards, Fred Coe
I am a 50 year old gastric bypass patient. Roul X complete gastric bypass was done in 2012. In 2019, I started having acute breathing problems. on investigation, it was found that there is no problem as regards, chest, ENT, heart. My serum calcium once came 12. but it was repeated thrice again and serum calcium was 9.5 to 9.7. ACE test is 36 within normal range (8-65). PTH intact is 36.7, also within normal range (15-65). Vitamin B12 was as low as 104 for which I am taking injections initially on alternate days for a week and then weekly since last 1 month. My urine calcium to creatinine ratio is as high as 0.45. Doctor says I have hyper calciuria. He has ordered for 24 hour urine calcium test. My chest CTis normal with 1 minor observation-“one pre-tracheal (12 X 8 mm) 15HU value and one sub carinal lymph node (17 X 8 mm) 12HU value seen”.serum ferritin is 20.20. RA total test is also within normal range. My bones specially spine aches a lot. I continue to have breathing difficulty. Doctor says it is all pointing towards sarcoidosis. what will be the treatment of this in case of gastric bypass patient. I shall be highly grateful for your suggestions at the earliest. I joined YOGA thinking it might help my breathing.
Hi Neelu, The main stone risk after rxy is increased urine oxalate, and I do not think the bypass is causing the high urine calcium – but that was a spot urine which can be remarkably unreliable. I suggest your physician order a kidney stone 24 hour urine from a reliable vendor as you get a lot of related valuable measurements that help in analysis. Litholink – a branch of Labcorp but you want to specify this name – is the best one. Normal serum PTH is not most ideal for the Dx of sarcoidosis. A serum calcitriol might be helpful. Regards, Fred Coe
1, 25 dihydroxy vitamin D3 report is 65.2 within normal range (19.9-79.3). I am going to collect urine for 24 hours starting tomorrow morning for 24 hours. I shall get the report in 5-6 days. What should be my line of treatment.
Hi Neelu, Your 1,25D is not remarkably high nor your PTH suppressed. I wonder if you have sarcoidosis or merely genetic hypercalciuria. It is a common genetic trait. A 24 hour urine will help as it gives a reliable number to gauge against proper normals whereas your spot urine is not reliable or usable in that manner as urine calcium depends a lot on food. If it is just IH, and you are not a stone former nor have bone disease, the finding may be of no clinical importance. Regards, Fred Coe
How does one know whether they are stone markers. what is a stone marker
Hi Neelu, I do not believe the term ‘stone marker’ is in this article or any other. I do not know what the phrase might mean. Fred
In my 24 hour urine calcium test- urine calcium came as < 5 mg/dL and urine volume was 6.7 litres in 24 hours. Report says that calcium 24 hrs urine could not be calculated as lower detection limit for calcium is 5 mg/dL. what inference should we draw from this report.
Hi Neelu, as an estimate, if your urine calcium were 4 mg/dl, or 40 mg/l, you would have 4 x 6.7 mg/d of urine calcium loss (268 mg). That is theoretically enough to raise stone risk but your urine volume is so huge no supersaturations are likely. If you can tolerate such high volumes, it is effective against stones. There are more elegant ways to prevent stones, in general, but perhaps this is fine for you. Regards, Fred Coe
I am a 51 year old woman who has known multiple kidney stones in both kidneys. They are stable. I have recently been diagnosed with osteoporosis. I have had numerous fractures. I had a 24 hour urine done with the calcium 324. PTH – 56. Serum calcium 9.8. I’m not sure what’s causing my bone loss. Could the existing stones be a reason why I’m spilling calcium in my urine? I’m currently taking Vit D and Calcium supplements. Doctor wants me to start osteoporosis meds but very scared due to side effects.
Hi Kelli, I presume you have idiopathic (genetic) hypercalciuria which is highly associated with bone disease and causes kidney stones. The high urine calcium could be from the rapidly progressive bone disease but the many stones were prior and that is most compatible with genetic high urine calcium loss. This article details IH and perhaps will be of value in terms of treatment. If you have had fractures your physicians are quite right to want to use bone active medications. Regards, Fred Coe
Thank you for the reply! I am currently trying to find a Doctor that an help manage my kidney stones and bone issues. I have been followed by a Urologist for the kidney stones. We have been ‘watching’ them for several years as they are in the lower pole of my kidneys and not causing any problems. Question I have is can the kidney stones be causing my bone density issues? Do they effect calcium absorption in the bones? Thank you very much for the information and response.
Hi Kelli, No. The stones per se are not known to affect bones. It is the underlying hypercalciuria that is common to causing stones and most likely also causing bone mineral loss. But other factors may matter for you given your age – menopause. Take a look at the article on idiopathic hypercalciuria. Also, ideal treatment for IH is also ideal for bone whatever the other causes. Regards, Fred Coe
I am a 32 yo female. I was diagnosed with osteoporosis in 2014 after being treated for a stress fracture that wasn’t healing and then having a DEXA scan done, and my bone density is gradually getting worse. No kidney stones ever. Low PTH: 13. High 24 hr urine calcium (500’s from 2014-2018, 400’s after doing the keto diet for 2 months this year, 200 after fasting for 2 days last week). 25mg thiazide had no impact on 24 hr urine calcium. Normal serum calcium levels, normal phosphate levels in urine and serum. High CTX (631, but dropped to 384 on 25mg thiazide). 25mg thiazide had no impact on PTH. Was treated with Depot Lupron in 2006 and 2012. Have been taking low dose birth control pills (Yaz and Yazmin) since 2006 to control endometriosis (except from 2009-2012). Everyone is stumped. Osteoporosis medications are not recommended for me because I am so young. The provider I’m currently seeing is working with me to “experiment” with different things to see if we can figure it out. Curious if you have any thoughts?
Hi Rebekah, You have a complex problem and need very expert help with it. Your PTH is a bit low, urine calcium remarkably high. I can imagine multiple causes, but that would be beside the point. I would advise you consult with physicians at a major center that deals with bone and mineral diseases. If you say where you live I would be happy to recommend some places. Treatment requires people figure out what is the cause of your bone disease. Feel free to email me directly: coe@uchicago.edu Regards, Fred Coe
Hello
I am a 69 y.o. Male who has had kidney and bladder stones off and on (fortunately mostly off) for about 25-30 years. Stones are mostly calcium oxalate. After my last stone episode my PTH was 173, and my blood calcium was 9.6. The PTH has jumped up from around 75 about a year ago but the calcium has always been under 10. My urine calcium was most recently around 300. A couple of years ago my vitamin D was quite low (around 13), and a supplement now has it around 50. i am inclined to blame everything on primary hyperparathyroidism and probably head for surgery, but the endocrinologist thinks it might be hypercalciuria and has prescribed 12.5 dose of hydrochlorothiazide to be followed in 6 weeks by a blood test. I am very tired all the time and also have insomnia which isn’t a helpful combination. A c/t scan from a few years back indicated medullary kidney, however no other scan interpretation reported that. Are the diagnosis of Hypercalciuria and use of the diuretic plausible.
Thanks for your very interesting site!
Hi Jim, The high PTH with normal blood calcium and vitamin D level are most consistent with secondary hyperparathyroidism from mild reduction of kidney function. At age 69 many have this a part of aging. Another possibility is low calcium diet – a very bad idea, and in some people able to raise PTH levels remarkably. Your physicians can look into the former, you into the latter. As for idiopathic hypercalciuria, PTH levels can run high, and that is possible. The diuretic – which can lower urine calcium – lowers PTH a bit, not raise it. I presume your blood is always fasting – it must be to make sense of serum calcium and PTH levels. Regards, Fred Coe
Dr. Coe – Thanks very much for your quick reply. It’s all very interesting and complicated! I have not knowingly been on a low calcium diet, but will read more about it, and my yes, blood was always fasting. Another perhaps pertinent recent lab value was Calculated GFR = 73, which I understand to mean my kidneys are not what they used to be! Given your comments and If secondary hyperparathyroidism is simply part of aging, then surgery on the parathyroid is not warranted, and the diuretic is logical to try in case idiopathic hypercalciuria is the culprit. The endocrinologist also ordered a baseline bone density assessment next week. Is my interpretation of your note accurate? Thanks again.
I believe so. Let me know what happened. Fred
Hi Dr. Coe:
A quick follow up after 2 months on hydrochlorothiazide. My PTH has dropped to 109, which is somewhat better and my blood calcium is pretty steady at 9.7. The physician is upping my dose of the diuretic to 25 mg from 12.5 and noted that things were moving in a good direction. My ionized calcium was slightly high at 5.45. They also discovered osteoporosis unfortunately, so we will see how that goes. Does idiopathic hypercalciuria still seem to be a likely issue here? Thanks!
Hi Jim, The normal serum calcium speaks against primary hyperparathyroidism; calcium ion can rise from the thiazide. IH is not a bad guess as it causes bone disease as well as stones. The slight increase of calcium ion and high PTH are just a bit worrisome, but the normal total calcium is reassuring. Is your serum albumin by any chance reduces – could give a lower total calcium. The high PTH has some cause – reduced kidney function, low calcium diet, low serum 25 D. I am sure your physicians are figuring that out. Regards, Fred Coe
Hi
I’m a 40 year old who has passed stones since 14 years old. Two doctors have prescribed thiazides because I have great blood work but calcium levels in urine are always in the high 300, low 400’s. I want to avoid drugs, but also avoid stones. Would you start a low sodium diet before trying the medication? I would like to try to fix this problem with diet changes if possible. Do you have any diet change suggestions?
Hi Allison, Indeed I would. Here is an article on your condition – genetic hypercalciuria. Here is another showing the interaction between diet and medications. Regards, Fred Coe
Hi Allison,
You must lower your sodium intake either way as the meds are less effective unless you don’t. You can talk to your doctor about doing dietary changes and redoing your urine collection to see how much that helps. Go here to understand how to implement dietary changes.https://kidneystones.uchicago.edu/the-kidney-stone-diet/
Best, Jill
I am a 39 yo female. Had a 24 hour urine test done 2 years ago. All was normal. Had one done last week and urine calcium was 665. All bloodwork normal. Passed 1 stone have 2 more. Have to go in for PTH test. I live in Seattle. Any recommendations on the best place to go to figure this out. Thank you.
Hi Meri, It seems remarkable that your calcium was normal and is not nearly 700 mg/d. I would suggest you use the university medical school as your resource as you have a puzzling picture. Of the physicians listed under kidney stones at UW this one seems most promising.Given telehealth is now more feasible, you can also seek consultation nationally. Regards, Fred Coe
Dr. Coe,
Thank you for your informative site! I’m 35 y/o female with history of stones since I was 17. Prescribed HCTZ off and on for years and low sodium diet. Recently started having dull flank pain off and on for months. Had multiple stones passed (confirmed on CT) back in March. My nephrologist did a 24hr urine and my calcium was 491. She suggested a low sodium diet again, which I’ve never had a high sodium diet, and possibly Chlorothiazide depending on my blood work. My serum calcium was 10.2 and PTH was 36 (2 years ago my serum was 9.9 with a PTH of 59). I don’t have a follow up with my dr until November. I am already on atenolol for HBP that I’ve been on for 2 years. I am uncertain whether I should be seeing an endocrinologist or waiting to see my nephrologist in November. I have had symptoms of hyperparathyroidism but I don’t think my dr’s think that could be causing my issues/symptoms.
Thank you for your help and informative website.
Dear Dr Coe,
Thank you for your work on hypercalciuria. I am 67ys old. I lived in East Africa as a child and contracted Schistomiasis (Bilharzia). It was treated in my 50s after evidence of on-going kidney issues. Since 2003 I have had stones which passed. (calcium oxalate crystals identified once in urine )
In April I had a right partial nephrectomy to remove a 20mm RCC.
Kidneys are currently good and function normally (>90). Blood tests and blood calcium are ‘normal’.
24hr urine 2.5l: Calcium 8.0 mmol/day. (N:1.2-7.5) Uric Acid 4.9 (N: 1.5-4.5) PTH 4.1 pmol/l (N: 1.6-9.3)
I have been prescribed daily treatment with low dose Indapamide (Thiazide type) and Allopurinol. If possible, I prefer to treat naturally with diet/exercise but will follow current advice if the best option.
Do you have any suggestions?
Hi Paul, You do indeed have high urine calcium, usually a genetic trait. The ideal approach is as in the article – reduced diet sodium and then a low dose of a thiazide type drug. Indapamide is fine, but without sodium restriction it is inefficient. Allopurinol is without purpose so far as you have told me. Regards, Fred Coe
Dear Dr Coe, Thank you for replying to my comment I have attended hospital once for a kidney stone. Since then I have found that drinking plenty of water – especially while working in summer – seems to limit, and be the solution to, occasional kidney discomfort.
Scans showed there was calcium associated with the RCC; all of which was removed during the partial nephrectomy. Can there be a causal relationship between high calcium urine and RCC development?
As you suggest, I’ll now try the Indapamide my doctor prescribed. I am currently on a fresh food – low sodium diet but I will work on further reductions.
I think the Allopurinol (100mg/day) is intended to reduce uric acid. Is that related?
Thank you again,
Paul
Hi Paul, RCC might have calcium associated but the mechanisms differ from stone disease. The high urine calcium is not likely to the RCC unless there was residual tumor – I gather this is not the case. The low sodium diet – 1500 mg sodium/day will lower urine calcium and perhaps obviate the indapamide. Allopurinol does indeed reduce urine uric acid but I cannot quite see its need given what you have told me. However your physicians know far more about you than I, and I would take their opinion over mine in all cases. Keep high fluids all the time. Regards, Fred Coe
I have had 9 cases of kidney stones and been in two surgeries.
Last stone in July 2020. The one before in February 2019.
One stone was analysed: calcium phosphate/calcium oxalate stone
Values from tests:
S-AFOS 110 U/l
fS-Pi 0.67 U/l
S -Ca-Ion 1.19 mmol/l
U -pH 5.5
dU-K 66.64 mmol/d
dU-Na 114.24 mmol/d
dU-Citrate 0.88 mmol/d
dU-Uraat 2.38 mmol/d
dU-Ca 9.71 mmol/d
dU-Oksal 24.8 mg/d
Amount of urine 2,3 liters.
12th February 2015 – M87.0 AVN (left hip) (idiopathic) (self healed fully 05/2015 after 3 months with cruthes) (Also Transient Osteoporosis suspected)
21st September 2015 – M87.0 AVN (right hip) (idiopatchic) (self healed fully 12/2015 after 3 months with cruthes) (Also Transient Osteoporosis suspected)
08/2016 Sudden loss of hearing in one ear without infection. (Noise injury pattern) (idiopatchic) Healed fully in two months.
I have had 6 bone fractures of which 3 times wrist.
My great grandfather, grandfather, mother and sister have had stones but no cause has ever been found.
Can there be a common factor for these diseases?
I have never had any medication in long time use and have no longtime diagnosis.
Should I have any medication to prevent stones?
Hi Thomas, You have mixed calcium phosphate calcium oxalate stones with very high urine calcium losses, possible bone mineral deficits and a family history of stones as well. Most likely you have genetic hypercalciuria. Your urine sodium is already reasonable but you would benefit from lowering it to 65 mEq/d from the present 114. Your physicians might want to consider adding a long acting thiazide like drug which lowers urine calcium. If your bone mineral content is low, you might benefit from bone active agents to stabilize it. Regards, Fred Coe
Hi Dr. Coe,
I am 52 years old. Diagnosed with thyroid cancer 17 years ago, had a complete removal. From 2011 through 2016 had numerous kidney stones and surgeries. Three years ago, they found a tumor on my last working paraythyroid gland. Had it removed. Had symptoms of hypopara, so it was reinsterted into my forearm. Still have very low PTH numbers but good steady calcium numbers in the blood. Was taking 2 calcium pills and my doctor put me on a hydrochlorothiazide due to high urine calcium but wasn’t working like he wanted, so he switched me to chlorthalidone and now my urine calcium is over 700+. This med is lowering my potassium and showing signs of that. I can’t find a true endo in the NJ area that deals with both. Very frustrating. Suggestions/comments.
Hi Duke, I believe I replied to you in your email. You have hypoparathyroidism with the unfortunate consequence of needing a high urine calcium to support your blood calcium. The CTD is not a bad idea, and low diet sodium would be a good addition to prevent potassium wasting and let the drug work better. Managing your condition is a tightrope act, so it needs a lot of adjustments. Regards, Fred Coe
Good Morning Dr. Coe,
I am a 41 year old male with chronic kidney stones (Stones began 17 yrs ago).
I produce predominantly, Calcium Phosphate, and I experience granular sand passing through my urinary tract 2-3 times per month with mild to moderate discomfort; sometimes immobility can occur. My urine calcium levels over the past 4 years have all been elevated and range from 340 – 460 mg/24 hrs. My blood calcium has been tested over the last 15 years and ranges from 9 – 10.1 mg/dl, with no obvious trends. My PTH has been tested twice in the last 2 years, resulting 27 & 45 pg/ml. I have had Vitamin D test as well. In the last 8 years they have ranged from 17.6 – 30.3 ng/ml. The frequency of stone passing over the last 17 years has gone from once a year to twice a month. I have been diagnosed with “leaky kidneys”. I also have been diagnosed with major depression (2006) & recently bi-polar (2019) & IBS (2007) by the VA hospital. Along with rapid stone passing, I find that I experience diarrhea (weight loss), soar throat / neck pain, & mild flu like symptoms. My doctor told me that the diagnosis of “leaky kidneys” is sort of the end of the road, but I am not comfortable with that, especially since I can’t tolerate diuretics due to the adverse side effects that I experience in direct sunlight while on the medication. Do you have any suggestions or recommendations for me, I live near White River Junction, VT.
Thank you in advance for any advice.
Hi Sean, I presume you have genetic hypercalciuria and ‘leaky kidney’ refers to our own work showing that the high urine calcium losses arise in part from reduced kidney calcium conservation. Phosphate stones reflect high urine pH, and unless due to one of your drugs appears to arise from high kidney ammonia production. If you cannot use thiazide very low diet sodium will reduce urine calcium losses. When diet sodium is low enough – 1500 mg Na or less a day – a very low dose of indapamide or chlorthalidone should be enough to reduce stones – the drug lowers urine pH as well as calcium. Sometimes light sensitivity varies and one of these two will work. Regards, Fred Coe
My son is 11 years old. He has a history of Very high calcium levels in his urine. He has been under the care of a nephrologist since he was 4 years old. His levels are very high. He has had a few passable stones. He also has a history of Nephrolithiasis. He has been on thiazides for 6+years. Is this just going to be his life (24 hour urines every Six months, ultrasounds/bloodwork every Six months? Or is research still being done? Any studies for kids? Thank you!
Hi Gina, He presumably has genetic hypercalciuria, which is generally present in about 1/2 of first degree relatives (you dad brothers sisters). Thiazide is often used in children and adults to reduce stones. From the physiology of the condition – no trials in children – one would favor a low sodium diet to reduce kidney calcium losses, and a high calcium intake to allow full bone mineral accretion by adult life. The low sodium high calcium diet is ideal if he can maintain it. Given low diet sodium, his dose of thiazide can be minimal – his physician is of course in charge of all this, and my notes are just that. The US ideal sodium intake for adults is 1500 mg/d, his could be that or lower. This is much lower than what the average US person eats, but our diet is judged too high in sodium. If his diet sodium can be reduced enough he may not need thiazide and therefore not need bloods every 6 months. Regards, Fred Coe
Dr. Coe,
My three year old son is under the care of a pediatric nephrologist and endocrinologist. One year ago he was found to have calcium in his kidneys. Further testing showed high blood calcium, 12.4 and high urine calcuim 500+, pth levels in normal range. Since then he has had a sestamibi parathyroid scan and ultrasound, three rounds of genetic testing and bone density tests all of which have come back “normal.” He was recently put on sensipar and with an adult dosage, his blood calcium is within range but calcium in urine is still above 500 on 24 hr tests. The most recent talk is about also prescribing his an additional medication in attempt to lower his calcium in his urine. I’m not really comfortable with him taking medicine without even knowing what is causing his problems, let alone the high dosage he is taking. It’s also been suggested that I meet with a surgeon to discuss removing his parathyroids. Again, this doesn’t feel like a wise for a three year old. This poor baby is having blood draws every ten days for months now with no end in sight. I’ve researched all over night after night and was interested in your article. Any suggestions on where to turn to for help??? I feel like I’ve exhausted our resources and am resorting to medicating a condition we don’t know the root of.
Thanks in advance,
Lindsey
Hi Lindsey, The high serum calcium and normal serum PTH does strongly favor primary hyperparathyroidism. Because sensipar increases sensitivity of the cell surface calcium receptor, it is presumed – in humans – and known, in experiments, to cause a remarkable reduction of renal calcium reabsorption in the cortical thick ascending limb which creates his ongoing massive hypercalciuria. Here is a reasonable review of PHPT. Surgical removal of some PT tissue may be his only reasonable choice. This review suggests a single adenoma is the most common cause of childhood PHPT – his massively high urine calcium and elevated serum calcium point to PHPT and the article to a curable adenoma. My concern is about the surgery – this should be done in a referral center for complex childhood diseases, not to slight excellent medical centers but to say that this is indeed a very challenging case and the first surgery is usually the most crucial one. Regards, Fred Coe
I have produced kidney stones since 16 years old. I am 39 now. I have been working with my urologist to try and reduce my calcium oxolate kidney stone production. In 2016 my urologist prescribed 20meq/day of potassium citrate and I have been mainly stone free. My 24 hour urine results from 2016 had my urine calcium at 261, so he decided to prescribe hydrochlorothiazide 25mg once per day in addition to 40meq potassium citrate. I just received my results from the most recent 24 hour urine expecting that the thiazide would help the urine calcium to go down but it went from 261 to 315. My litholink results are recommending to increase the dose of the thiazide. Will this help even though after going on the thiazide my urinary calcium increased? Here are the rest of the results from the litholink 24 hour urine analysis.
Urine Volume: 2.57
SS Caox: 4.5
Ca: 315
ox 24: 22
Citrate: 364
ssCaP 2.05
ph 6.76
ssUA .04
UA 24: .261
Na 24 :81
K 24: 44
ca 24/kg: 6.3
ca 24/cr24: 339
I went from 20meq/day of potassium citrate to 40meq/day to try and increase the urine citrate, but as a result my ph went from 6.45 to 6.76 (when I wasn’t taking potassium citrate my ph was 5.89) and ssCaP went from 1.28 to 2.05. Is this a problem that will lead to Calcium phosphate stones? Litholink is also suggesting I increase my potassium citrate dose but I’m wondering if its the right thing to do since my PH and ssCaP are already showing in the high level? I am finding it really difficult to find the right balance. My main concern right now is how to get the urine calcium down because of the stone risk and bone issues it can lead to. So will a higher dose thiazide be good to try or is there something else I can do to get the urinary calcium down? I can’t get in to discuss the results with my doctor until next month, so would appreciate your opinion so I can find the best approach with him at my appointment. I really want to avoid producing any more stones!
Hi Diana, You presumably have genetic hypercalciuria – normal blood calcium, high urine calcium, no known systemic diseases, and urine SS control is inadequate despite high volume and low sodium – 80 mEq/d. You are right – pH will go up with K citrate, and citrate in urine is low. Urine potassium is 44 which is odd as you take 40 mEq of potassium citrate and also must be eating at least 40 – 60 mEq of potassium, so perhaps you were not taking the med or are potassium depleted. I might suggest stopping the potassium citrate in favor of potassium chloride, perhaps a higher dose, and a longer acting thiazide like chlorthalidone – 12.5 mg. See what your physician thinks as she/he is totally in charge and I a mere bystander. Regards, Fred Coe
My mother has high calcium in blood and low in urine. Her thyroid tests came back normal. What could she possibly have? Thanks for your help.
Hi Tiffiny, High blood and very low urine calcium can be from a benign familial trait: familial hypocalciuric hypercalcemia. It is not a stone forming state, and does not require treatment. Alternative reasons are primary hyperparathyroidism + either kidney disease or very severe vitamin D deficiency. Incidentally, I noted that on Google all of page 1 and much on page 2 are advertisements paid for to put commercial material up front. The article I linked to is a fully reliable source. Regards, Fred Coe
Hello there. I am a 37 year old female who has had three stone episodes since last March. I was only able to get one of these analyzed, and it was 55% oxalate and 45% carbonate apatite. I have 3 additional 3-4mm stones plus a handful of punctate stones in my left kidney. The right kidney is free and clear of stones and continues to show so on all my scans, but has had unexplained mild hydronephrosis for several months (no stone or signs of obstruction anywhere, but I have yet another u/s next month so see if there’s been a change in swelling in the last 6 weeks). Anyways, a little background information. I had a 24 hr urine test done while not passing any stones and here were the results:
Cystine screening: negative
Vol 24: 2.06
SS CaOx: 4.21
Ca 24: 306
Ox 24: 19
Cit 24: 830
SS CaP: 2.12
pH: 6.339
SS UA: 0.36
UA 24: 0.707
Dietary Factors:
Na 24: 125
K 24: 58
Mg: 119
P 24: 0.969
Nh4 24: 45
Cl 24: 135
Sul 24: 33
UUN: 9.63
PCR: 0.9
Normalized values:
Weight: 89.8
Cr 24: 1363
Cr 24/Kg: 15.2
Ca 24/Kg: 3.4
Ca 24/Cr 24: 225
My urologist has put me on 12.5 hydrochlorothiazide to try and help manage the high calcium in my urine. They tested me for hyperparathyroidism but my serum calcium, parathyroid, thyroid, and vitamin D all came back in normal range.
Was just curious to see what your thoughts were. Thank you for your informative articles!
Hi Alma, You do have hypercalciuria and I presume it is genetic – here is my best article on it. Your stone has a lot of phosphate in it, so you are close to the calcium phosphate variety of stone former, making prevention specially important. I surely like the dose and type of thiazide but would suggest lowering diet sodium as well, to below 2000 mg/d so the drug will work better and you will waste less potassium in your urine. I would avoid potassium citrate and use potassium chloride should you need potassium replacement, as your stones form in alkaline urine. Regards, Fred Coe
Hello; I am a 68 year old stone former with IH. I also have 4 fully formed kidneys with 4 separate ureters emptying into the bladder. My most recent 24 hour urine collections found my urine calcium at 340 (it was 382 two years ago), sodium at 87, and citrate excretion at 783. I had only 1.7 liters after the 24 hour collection. Calcium Oxylate crystal 2.42, Brushite Crystal -0.63, Uric acid crystal 1.42, sodium urate crystal -0.50. Blood test showed Parathyroid at 20 pg/ml, and blood calcium of 10 mg/dl. I had a 4mm stone removed two years ago, and now have one 3mm, and two tiny 1mm, all in my two right kidneys, and a tiny plague deposit on one of my left kidneys. First, do the numbers suggest parathyroid issues might be possible? I can’t take the thiazide treatment because of an enlarged prostate and the already existent need to pee constantly. I am now up to about 2.5 liters of water and crystal light a day and frequency of urination has increased significantly. Add a diuretc and I believe I would have to be homebound. So…will preventing supersaturation with > 2 liters of fluid (one liter of which is crystal light) and/or potassium citrate pills, getting my citrate level much higher, still prevent stones even with the IH?
Hi Robert, Very high urine calcium, and citrate, low volume. You do not say what the stone was made of – was it calcium based??
The single calcium of 10.0 is at the top of the normal range. Possibly you have hyperparathyroidism, but the only way to tell is whether fasting morning serum calcium values are above normal or not. Usually when in doubt I do three, on three separate mornings, assuring fasting. If normal I presume you have genetic hypercalciuria. Treatment depends on the diagnosis that is not as yet certain. The presence of 4 kidneys with presumably thin ureters makes proper diagnosis even more important. If possible you would be best off getting your care at a university based referral center for kidney stones. As for urine volume, diuretics do not increase it, they simple alter the relationship between diet sodium intake and urine sodium loss. Long acting thiazide type diuretics have no sudden urine flow increase such as it seen with short acting hydrochlorothiazide. Your prostate problem needs fixing so you can maintain at least a 2.5 liter daily urine volume. Regards, Fred Coe
Hi I am a 70 year old male who has had kidney stone issues for most of my adult life
I was diagnosed with Idiopathic Hypercalciuria many years ago I have had in excess of 60 stones. sometimes as many as 10 at a time.
Lithotripsy 6 times, passed many, even having open kidney surgery to remove very large stone from the renal pelvic cavity.
I do suffer with osteoporosis and frequent UTI Have tried hydrochlorothyaside but to no avail I still get frequent stones.
Could this be related to parathyroid.
Hi David, Genetic hypercalciuria causes stones and bone disease. Primary hyperparathyroidism does seem related to IH, and certainly is very important to exclude or diagnose and cure. Here is a good overview of an evaluation that gets all causes. Be sure you have been evaluated properly, and look into the hyperpara article about how bloods must be collected. Regards, Fred Coe
Hi Dr. Coe, I am a 30 year old woman with known diabetes. Several years ago, I was found to have high serum calcium (as high as 11.1, which repeated several months later was the same, and by a different lab). My PCP did a work-up and found my PTH at the time to be 13 (at the same time as the second Calcium of 11.1.) My PCP then sent me to an endocrinologist — since then my serum Calcium has been in the mid-9’s and PTH in the 40s; because of this, she wants to wait and watch to see what happens. However, she did a 24 hour urine calcium as part of the initial work-up and it resulted as 507 with a 24 hr Creatinine of 1470, pH of 2 (total volume = 1.5L); called it a fluke and retested this year — 24 hr urine calcium now 796 with a 24 hr Creatinine of 1456, pH of 6 (total volume = 2.6L). We did scans (4D CT and u/s) and found no evidence of primary hyperparathyroidism. Of note, my Vit D has been very low for years — the lowest was 15, most recent was 25 on 800 IU/daily. My endo wants to send me to a nephrologist to evaluate for renal tubular acidosis (though I have my doubts as my urine has not been alkaline). I have not had kidney stones to my knowledge, though have not had a kidney u/s to confirm the lack of in the setting of the high urine calcium. I have had joint pain (right hip specifically), muscle weakness, headaches, and marked fatigue for several years now; have a history of high-normal Calcium (noted, but not investigated until the 11.1 values) and gastroparesis pre-dating my diabetes; I also have a history of irregular, light, long periods for which I am currently on the birth control patch (and was previously on Depo). I have not had a bone density scan. My new PCP wants to go down the route of a sleep study for the fatigue and a rheumatologist for the joint pain. I’m concerned that all of this could be connected and that I’m gonna go through a bunch of tests and specialists with no results. Any thoughts on how to piece this together? Could it just be the diabetes (last A1C was 7.2)? Thank you for any thoughts you can offer! -Leigh
Hi Leigh, The very high urine calcium, low PTH, and low 25 vitamin D suggest increased levels of active vitamin D (1,25D) that can be measured. In general 1,25D is not specifically high in diabetes, but needs checking. Both type 1 and type 2 diabetes cause bone disease, I do not know which you have, but if it is type 2 then it is the relatively less common MODY diabetes in which high urine calcium may be more marked. Your diabetes is not well controlled. I would think better diabetic control would be your best alternative. As for RTA, that makes no sense – bone mineral loss or excess 1,25D is driving the high urine and intermittent high serum calcium levels. You should have a BMD scan, but the bone disease of diabetes is not simple mineral loss, but a loss of normal bone toughness due to glycation of bone collagen and myriad other issues. Again that points to better diabetes management. So, yes, I think the diabetes is causing the whole picture, it is either Type 1 or MODY, and you would benefit from expertise specifically directed to the problem of diabetes and bone. Regards, Fred Coe
I have severe hypercalciuria and hyperoxaluria and have been diagnosed with nephrocalcinosis. My urine is light in color but always cloudy with visible particles. Could this be caused by the hypercalciuria? I also have a burning sensation at times when I urinate. Could this also be attributed to the hypercalciuria?
Hi Emmie, Passing crystals means that you are forming crystals all the time. You need to collect the crystals and find out what they are, then using 24 hour urine samples figure out what is causing the supersaturation that keeps the crystals forming. I presume it is the high urine calcium, maybe the oxalate, and both have causes that can be modified so the crystals stop. These crystals are the basis for stones and possibly the nephrocalcinosis. Regards, Fred Coe
Mr. Coe,
For those of us who found high Urine Calcium in our 24hr Urine tests. Then found we had low bone density via a Scan (Not Osteoporosis though. Just lower than expected density). If a follow up 24hr Urine test brought the Urine Calcium back into normal by reduced Sodium and protein intake. And increased dietary calcium intake. Can we expect a follow up Deca scan to show our bone density has increased? If so, how long could that take for bone density to increase?
All of my 24hr tests are below. I typed by hand so hopefully there are no typo’s or numbers out of place. The latest test in December of 2020 shows how I corrected things by lowering sodium and protein. Drinking 2 tablespoons of pure lemon juice throughout the day. Using 2 packets of Litholyte per day. And increasing dietary calcium with milk and sardines. The results in the December 2020 test are quite dramatically different from my previous tests. I think you’ll find. My blood serum levels of all electrolytes have always been normal. I could also include those tests in a follow up post if it would be helpful.
Of note. My kidney stones started in early 2015. About 1.5 years after having my colon removed due to Ulcerative Colitis. I have an ileostomy. Immediately after colon removal I began adding a lot of extra salt to my food. As I was told I would need extra sodium going forward. I did not however, heed the advice to drink a lot more water. I didn’t feel any effects so I figured I was ok drinking the amount I’d always drank.
In 2015 I started noticing a burning when I would pee. And started seeing what looks like hundreds of tiny crystals in my urine. Urologist appointment confirmed kidney stones. I was told to drink more water and add some lemon to it. I began drinking 1 gallon of water a day and never stopped since then.
My urine tends to get very dark. Very quickly. If I do not drink about 1 gallon of water through out the day. Reguardless of how active I’m being. I believe this is due to not having a colon.
I have maintained about 1 gallon of water a day since my diagnosis of kidney stones in late 2015. In spite of that. I would still get the stones. I’ve had 4 total 24hr Urine tests since 2015. Results below. Again, it seems I can correct things with the diet changes mentioned above. I am most concerned with adding bone density back. Since my scan earlier this year showed low density. I am 40 years old. 6ft 150lbs. Very active and wish to not be limited in the future via weak bones. I am also curious what your thoughts are on Calcium supplements from Microcrystalline hydroxyapatite. Any thoughts are appreciated.
9/24/2015:
Urine Volume – 2.57
SS CaOx – 5.56
Urine Calcium – 280
Urine Oxalate – 39
Urine Citrate – 545
SS CaP – 0.32
24 Hour Urine pH – 5.429
SS Uric Acid – 1.50
Urine Uric Acid – 0.758
Urine Sodium – 62
Urine Potassium – 101
Urine Magnesium – 151
Urine Ammonium – 63
Urine Chloride – 104
Urine Sulfate – 81
Urine Urea Nitrogen – 19.40
Protein Catabolic Rate – 2.0
Urine Creatine – 2250
Creatine/kg – 33.1
Calcium/kg – 4.1
Calcium/creatine – 125
9/07/2016:
Urine Volume – 4.32
SS CaOx – 2.70
Urine Calcium – 397
Urine Oxalate – 26
Urine Citrate – 603
SS CaP – 0.35
24 Hour Urine pH – 5.805
SS Uric Acid – 0.60
Urine Uric Acid – 0.870
Urine Sodium – 124
Urine Potassium – 63
Urine Magnesium – 137
Urine Ammonium – 92
Urine Chloride – 152
Urine Sulfate – 88
Urine Urea Nitrogen – 19.77
Protein Catabolic Rate – 1.9
Urine Creatine – 1881
Creatine/kg – 26.8
Calcium/kg – 5.6
Calcium/creatine – 211
8/27/2020:
Urine Volume – 2.80
SS CaOx – 5.33
Urine Calcium – 448
Urine Oxalate – 34
Urine Citrate – 370
SS CaP – 0.36
24 Hour Urine pH – 5.233
SS Uric Acid – 1.91
Urine Uric Acid – 0.847
Urine Sodium – 109
Urine Potassium – 65
Urine Magnesium – 196
Urine Ammonium – 110
Urine Chloride – 158
Urine Sulfate – 76
Urine Urea Nitrogen – 21.44
Protein Catabolic Rate – 2.2
Urine Creatine – 1779
Creatine/kg – 26.5
Calcium/kg – 6.7
Calcium/creatine – 252
12/04/2020:
Urine Volume – 4.16
SS CaOx – 2.00
Urine Calcium – 167
Urine Oxalate – 30
Urine Citrate – 830
SS CaP – 0.59
24 Hour Urine pH – 6.337
SS Uric Acid – 0.15
Urine Uric Acid – 0.574
Urine Sodium – 94
Urine Potassium – 57
Urine Magnesium – 137
Urine Ammonium – 33
Urine Chloride – 89
Urine Sulfate – 29
Urine Urea Nitrogen – 9.10
Protein Catabolic Rate – 1.0
Urine Creatine – 1561
Creatine/kg – 22.9
Calcium/kg – 2.5
Calcium/creatine – 107
Hi Douglas, Ileostomy is causing your problems, and perhaps ulcerative colitis before it. The colitis can reduce calcium absorption and reduce bone mineral density, and steroids’ – if used – can much damage bone. Ileostomy causes loss of water, sodium, and alkali resulting in an acidic low volume urine that produces uric acid and calcium oxalate monohydrate stones. You have the sodium alright but needed sodium bicarbonate as a reliable alkali. You do not mention the stone analysis but they will be more or less as I said. The lemon juice is always inadequate given ileostomy. Potassium citrate is often not well absorbed. Sodium bicarbonate tablets are OTC, cheap and reliable – as in the article. The stones persist because the dark urine episodes cause showers of crystals. The cause of your bone disease may be complex given your underlying colitis in the past, and may require bone directed drugs beyond the scope of this site at present. Regards, Fred Coe
Thankfully I was never on steroids for very long. They never helped me! I have never had my stones evaluated. They are always very tiny, orange/reddish, and lots of them. I also have never experienced a full on stone with the crippling pain. Thankfully. Always just the gravel. I’m going to try and collect some though and have it evaluated. Good to know about the Potassium citrate not being well absorbed without a colon. The Litholyte product I’ve been using is 7.5 mEq Potassium Citrate and 2.5 Sodium bicarbonate. Sounds like it won’t be of much use. I came across the article you linked a few days ago. And already ordered some Sodium Bicarbonate pills from Amazon. I believe the brand is called “Rugby”. I’ll be doing another 24hr urine test to see how the pills change things. Also, I’ve been Colitis free since 2013 when they removed my colon. No other issues there. No other disease which is great. So, my hope is the bone density issue was caused via the last 7 years of eating too much sodium and not enough calcium. Hoping I can reverse the bone density with diet. There is also a possibility that I just have lower than normal bone density. And it may never be an issue. So say’s the Endocrinologist. My density levels were not alarmingly low. Just lower than normal. A follow up scan next year will tell us more. Thank you for all your work on this website and for answering my questions. It’s incredibly generous and very helpful.
Hi Douglas, the orange – red marks then as uric acid and alkali will prevent them as urine pH rises near 6. OTC sodium bicarbonate is very inexpensive, and any pharmacy will have it. As for your bones, loss of mineral is well known in patients with inflammatory bowel disease. Here is a recent article. Certainly reduced calcium absorption can play a role, as can inflammation, but I think the bone disease is a complex problem and treatment not necessarily simple – it is beyond the present scope of the site so I have not written about it. Best, Fred
Thank you for the information on how my ileostomy itself may be the cause of my low bone density. While incredibly frustrating that none of my specialists seem aware this commonality, I am grateful that you’ve illuminated the topic for me. It’s also a bit depressing to think I had solved the low bone density issue via your advice surrounding my high urine calcium (lowering sodium, increasing dietary calcium), only to find out it’s probably something else causing it (ileostomy). Or maybe a combination of the two. Either way I am no doctor. Although this has become a full time job aside from my actual job. Trying to put all these pieces together. I am meeting with a new Endocrinologist next month. And I’ll be bringing a copy of the paper you linked. My hope is a consistent increase in dietary calcium and vitamin d, along with my new lower sodium approach that brought my high urine calcium into a normal range, will add some density back to my bones after some time.
Hi Douglas, If there is a lot of bone loss, your physician may want to employ a more modern bone directed therapy. But certainly your bone loss cannot be separated from the effects of ileostomy and inflammatory bowel disease. Fred
Sorry, one last thing. If you look at my last 24 test I did raise my Urine PH above 6. I thought I did this via the Litholyte packets and lemon juice. But as noted in my last comment. Litholyte is 7.5 potassium citrate and 2.5 sodium bicarbonate. If potassium citrate is usually not well absorbed by people with an ileostomy than perhaps I am an outlier? I am not sure how else to explain the success of getting my Urine PH above 6.
Hi Douglas, Potassium citrate pills are not well absorbed because of reduced GI transit time, but sodium and potassium bicarbonate dissolved in water will be absorbed early in the intestine and act as alkali. So you are as expected. Fred
Gotcha, so I should not worry about the sodium in Sodium bicarbonate pills raising my urine calcium?
Hello Dr. Coe –
I am a 38 year old female, exercise regularly, eat a mostly vegetarian diet, and have always maintained a normal weight. I have been passing kidney stones for almost 10 years. I had a stone analyzed recently:
Calcium Oxalate Dihydrate (Weddellite) 30%
Calcium Oxalate Monohydrate (Whewellite) 35%
Carbonate Apatite (Dahllite) 35%
I’ve repeatedly had high PTH, slightly low vitamin d, normal blood calcium but high calcium in 24 hr urine (364 mg). My calcium was 9.7, 9.4 and then 9.1 after taking calcium and vitamin d supplements for a month as directed. My PTH was 65.4, 81.5 then 89.2 after taking calcium and vitamin d supplements for a month as directed. My vitamin d was 24 and then 26 after taking supplements for a month.
I am wondering about early primary hyperparathyroidism vs hyperpcalciuria. My doctor wanted to prescribe hydrochlorofiazides, but I would like to avoid that. I took an rx with a diuretic years ago and it gave me terrible headaches. Plus I don’t want to take hydrochlorofiazides for the rest of my life with the side effects I’ve read about. Right now my dr is going to have me repeat 24 hr urine and blood work again in 3 months. Are there other tests or steps you recommend?
Thank you.
Hi Kate, Normal blood calcium and high PTH is secondary hyperparathyroidism, perhaps related to your low 25d levels. I would replete my vitamin D to a higher range of perhaps 35-40 and be sure that serum calcium remained normal as I suspect it will. You seem to have idiopathic hypercalciuria, and treatment can be with low diet sodium and avoidance of excessive protein loads or refined sugar above US guidelines. As your stones have considerable phosphate, I would advise considering a thiazide if urine calcium and calcium phosphate supersaturations cannot be brought into the normal range with diet and – of course – at least 2.5 l/d of urine volume. Regards, Fred Coe
Hello Dr. Coe,
I am a 5’4″ 112 lb. 52-year-old post menopausal vegetarian (I eat milk and eggs) woman who has been diagnosed with osteoporosis of my spine and osteopenia of my hips. After ruling out other causes of osteoporosis, my doctor diagnosed me with idiopathic hypercalciuria (I have never had a kidney stone). My most recent 24-hr urine result, after being on 25 mg HCTZ for several months, was 456 mg with 4800 mL urinary output. Blood levels of calcium, potassium, vitamin D, and sodium were WNL. The only other medication I take is 88 mcg of levothyroxine for hypothyroidism, and I began supplementing with 1300 mg of calcium carbonate, spilt between 2 doses, once I was diagnosed with osteoporosis. My doctor increased my HCTZ dosage to 50 mg about 2 months ago, and I have a repeat 24-hour urine test in about a month. I’m drinking a lot of water and taking in sodium trying and keep my blood pressure above 100/60, and I’m in the bathroom all the time and generally feeling punk. Will I need to continue increasing my HCTZ dose to get my calcium excretion under control? Are there dietary adjustments I can make? An alternative medication? I wonder if the increased sodium I’m taking in to try to keep my blood pressure up is interfering with the HCTZ. Any information you could provide would be greatly appreciated!
Thank you,
Emily
Hi Emily, You do have a very high urine calcium and may have idiopathic hypercalciuria or rapidly progressive osteoporosis – or both. Your BP went down with the OHCTZ so you are adding diet sodium but that will more or less counteract the effect of the diuretic on urine calcium. I would suggest stopping the drug and lowering your diet sodium as much as possible – below 1500 mg/d of sodium as a start – which will not cause undue fall in BP and may do more for your urine calcium losses. Most probably you will want to try a bone directed medication – like a bisphosphonate – to see if reducing bone loss moderates your hypercalciuria. Yours is a complex situation so what I have said may not be correct in that I do not know all the facts about you, so my remarks are by way of thoughts your physician might consider useful or perhaps beside the point. Regards, Fred Coe
Dr. Coe,
The different diseases you mentioned that may cause this, are they all diagnosed by a blood test? If not, could you list how they are diagnosed? Thanks
Hi Darin, Mostly by 24 hour urine testing. Here is my best on evaluation. Fred
Dr. Coe, I have a hypothetical question……..if someone were a vegan for years and never ate animal protein or any dairy products and this person were to eat these, which one would raise the urine calcium the most? Let’s say they started eating either 1000-1200mg of dairy calcium per day or say around 60g of animal protein from meat or eggs per day, which one would show the most marked urine calcium increase? We’ll assume they don’t take any calcium supplements already.
Hi Lisa, I have just the article for you! Here are the three proteins, dairy, animal, plant in relation to stone risk, and a lot about effects on urine calcium. Here is my other one on protein and bone. The protein story is so interesting, I spent a lot of time on these articles, and it is no nice to have someone come along who is interested. Regards, Fred
Hi, I have severe osteoporosis in my spine and osteopenia in my hips. My initial 24 hour urine testing prior to intervention (Teriparatide), showed that I had hypercalciuria. The rheumatologist classified it as idiopathic in nature since other testing (PTH and blood calcium was deemed in the normal range). I was treated with low dose thiazides. After 1 year of treatment with Teriparatide, I repeated the 24 hour urine testing which showed a significant increase in my urine calcium level and my thiazide medication was doubled. Now after 2 years on Teriparatide, I had minimal improvement in my bone density levels (still at a -3) and my doctor is recommending a different bone drug. Is there a way that I can find out what it’s causing this leaching of calcium into my urine? I am so disheartened by this disease and searching for answers. ….. what additional reading should I be considering? I developed osteopenia at an early age (early 40s) and took 2 other bone drugs prior to Teriparatide. I maintained my bone density until for over 10 years and then a repeat DEXA showed significant change and severe osteoporosis in my spine (-3.3). At that point I had further testing, including the 24 urine test and discovered the high levels of calcium in my urine.
Hi Teresa, You may have genetic (idiopathic hypercalciuria) or even primary hyperparathyroidism but things are now so complex one cannot find out. I would stop the thiazide, stop the teriparatide, wait a few weeks and do a few fasting morning serum calcium + PTH + 1,25 and 25 vitamin D blood samples as well as another 24 hour urine to be sure the serum calcium is really normal and that there is not some other underlying issue and to assess urine calcium sans drugs. If there is nothing wrong (even slight increase of serum calcium, or a very high 1,25D or – possibly overlooked 25 D you will indeed be better off moving to another bone drug. I should say this is by way of an outsider comment as your case is very complex and I do not know enough details to be of primary medical value to you. Regards, Fred Coe
Hello Dr. Coe,
Is it possible or very common that a person can have high urine calcium, high PH, and a lower citrate in the urine that would sound like a calcium phosphate stone former but this person never get stones? I don’t know anyone like this but was just curious if people could have abnormal 24 hour urines and I don’t mean just one sample but maybe in a research study over time and never create stones.
Hi SB, Indeed. In the epidemiology studies from which we have derived the relationships between new stone onset and urine chemistries a given value will increase risk but certainly many escape. The same with car theft – part in a bad neighborhood and your car may never he stolen even though the prospective risk is high. This is a reason for some of my smarty-pants friends you took lots of chances during COVID and ‘…never had any problems, but had a lot more fun than you did.’ They never changed anything, went out for supper as always, flew around if planes were flying, and never got COVID. Same mathematics as your question poses. Best, Fred
Dr. Coe,
My situation is very similar to Teresa’s, above, having both osteoporosis in my spine, normal hip DXA and hypercalciurea, but although my endocrinologist was originally recommending Teriparatide, she is now having second thoughts because Teriparatide increases urinary calcium (although for completely different reasons than hypercalciurea) and can increase the likelihood of kidney stones. Do you have any knowledge/experience/comments on this? (Especially whether Teriparatide is a poor choice for someone with hypercalciurea) The endocrinologist indicated she tends to be more cautious than some others and many endocrinologists aren’t especially concerned about this.
Hi Barbara, I presume you are not a stone former but have high urine calcium observed during evaluation for bone disease. Teriparatide acts like parathyroid hormone and when given intermittently produces mineralized new bone. One uses it when fracture risk is high because of bone mineral loss as opposed to bisphosphonates that simply reduce bone mineral loss and stabilize bone. The choice depends on the bone disease. Stone risk can be increased but one can mitigate it by a low sodium diet – 1500 mg/d which helps reduce urine calcium. Regards, Fred Coe
My daughter, developed a stone at age 19. She is now 22 and had several 24 hour urine draws. She has done the 24 hour collection and is at 300 mg/d of calcium without HCTZ. With HCTZ, she gets down to 200 mg/d. The thing I don’t understand is part of her treatment has been to take calcium supplements. Both of the cited numbers are WITH the calcium supplements. Isn’t it worth doing a 24 hour urine draw without the supplements? I have a difficult time finding anything that suggests taking calcium supplements for treatment of calciuria. Her levels have decreased significantly since the first finding of her stone. Could they be continued to be elevated as a result of the supplementation?
Hi Heidi, Did she not have her baseline 24 hour measurements made without treatment?? And why supplements? Usually one wants to eat 1000 mg/d of calcium as food, the US recommendation for all Americans. Supplements, if used, need to be with the larger meals to avoid surges of urine calcium. A proper diet sodium is 1500 – 2000 mg, calcium 1000 mg, and I would do ‘baseline. measurements on that. If her urine calcium is indeed high with that food plan then she does indeed that an underlying problem with kidney calcium handling and might benefit from lowering sodium intake to the low end of 1500 mg. If that is not enough then one uses thiazide. Here is a reasonable statement of the kidney stone diet. Regards, Fred Coe
Hi Dr. Coe,
Thanks for this informative read! I have been experiencing issues for a year, and after a year of bouncing from doctor to doctor and a CT indicating kidney calcification I’ve finally landed in with a team of nephrologists.
I am in my 30s, and female. My 24 hour collection indicated a result of 435 for urine calcium and I was flagged for “extreme Hypercalciuria”.
They checked PTH and all is normal so next is vitamin D 1,25. I do have rheumatoid arthritis but it is well controlled and I cannot imagine is contributing.
I’ve been put on a diuretic and will undergo another 24 hour collection. I’m also drinking lots of water and adding lemon as recommended. I don’t have a special diet and I eat pretty balanced.
I’m feeling a bit desperate to get this resolved since it’s been a year now. I also get mild disorientation/brain fog, pelvic/abdominal pain, and I can literally see the “flakes” in my urine almost every time I use the bathroom, eek.
Do you have any thoughts or recommendations for me? Is there hope to make this stop and not be tied to medication forever?
Hi Jane, You are very hypercalciuric! I presume you have had a complete serum and 24 hour urine evaluation, stone analysis is known (Calcium phosphate? Oxalate?). The 1,25D is good, perhaps you have a gene defect in your phosphate transporter (Npt-2c) or CYP24a1 (the enzyme that degrades 1,25D). Many possibilities. Given nephrologists are working on your behalf I am sure they will come up with the right answer. I would be sure that testing is not influenced by treatments (I mean your physicians should be sure). Regards, Fred Coe
Hi Dr. Coe – I really appreciate your response. The 1,25D came back all normal. I have indeed had the 24 hour collection done via Litholink and am due to repeat this again soon now that I’ve been on the diuretic for a month+, but I am not sure about the “complete serum” and if that is a part of that. I see on the labcorp form it asks if a “serum blood draw” was requested and I have not had that done – so I am guessing I haven’t had the serum done unless that’s a basic panel? In which case, I have had a BMP run.
The nephrologists seem to indicate they believe this is genetic/idiopathic even though I do not know anyone within my family/extended family that has had extreme Hypercalciuria – only my grandma had kidney stones on one or two occasions.
This is probably difficult to answer without having my full background, but do patients with IH who are put on diuretics to manage ever get the opportunity to get OFF of the diuretics? Or is that not really a possibility?
Also, do you think it’s worth exploring the genetic testing to confirm it is truly IH and not something else causing this?
Adding to my reply – if helpful:
SS CaOx: 6.17
Urine Calcium: 435
Urine Oxalate: 21
Urine Citrate: 436
SS CaP: 1.89
24 hour PH: 6.1
SS Uric Acid: 0.37
Urine Uric Acid: 0.493
Do you do virtual consults/appointments? May be interesting to hear what you think and see if it aligns with what we’re doing so far.
Hi Jane, I apologize for the lateness, a number of comments were buried last winter. Your high urine calcium is the obvious cause of calcium stones and probably is already being treated by your physicians. If not indeed I do virtual consultation. Regards, Fred Coe
Dr. Coe, I have Ca Phosphate stones and have only changed 3 things and wondering if any of them are the cause of increase in urine calcium, citrate, phos, and SS. I am 54 went off birth control pill after 36 years of use 2 months prior to my recent 24 hour urine, I added one half extra banana and one half cup of milk to my diet daily. I drink large amounts of water and low sodium diet as you can see.
Urine calcium 270-319, citrate 359 to 561, sodium 61 to 57, volume 3.41 to 4.09, phos 696 to 806, Brus SS 2.28 to 2.90%, PH 6.8 to 7.2
Would the small amount of extra potassium in the milk and banana increase my citrate that much and PH? I know citrate is suppose to be higher but also know that increases PH and SS also. I have read some articles that estrogen use can affect urine calcium and wondered if recent going off BCP could affect all these. Will see urologist in the near future for follow up but so far he has not wanted to put me on thiazides to prevent new stones.
Hi Carol, Your studies have the very mark and make of calcium phosphate stones – in urine, high calcium, and pH, and low citrate. I gather you were forming stones while on BC pills, given the times you mention, but it is true that stopping BC pills may increase urine loss of bone mineral. My surprise is the high CaP SS with a urine volume near 4 liters, even though pH is near 7 and calcium near 300 mg. Is it a Litholink study?? I cannot make any recommendations lacking so much information, but if the stones have been forming for some time perhaps thiazide is the lesser of two evils. Regards, Fred Coe
Dr Coe, no this is not Litholink. I work for a hospital and this is what they use and send to ARUP Lab from my understanding. They don’t test ammonia or sulfate and all my other parts of this urine are normal. I started getting stones late 40’s and was on BC pills many years. I certainly hope they didn’t cause them. My fasting blood calcium is always below 10. If you increased your dairy by 80-100 mg a day how much would that increase your urine calcium if you changed nothing else? I already keep my sodium around 1200 mg a day, calcium 800 mg a day all from food, added sugar around 15 g a day, and my protein around 60 g a day, not much meat. I eat 8 servings of fruit and vegs a day. I feel that adding dairy will just end up in the urine and not helping. How low is safe to go with sodium intake? My urologist doesn’t seem to want me to take thiazide, said side effects from those but I’m very healthy otherwise and hope next month we can revisit this.
Hi Carol, I could not find yout original comment on this article, but I presume you have elevated urine calcium excretion – this site platform has limitations. Given your information I have to presume you have genetic hypercalciuria. In this condition urine calcium is higher than normal at any diet calcium intake and bone mineral can be lost. If thiazide is not acceptable to your physicians then very low diet sodium is next best – 1500 mg/d. This should permit a reasonable calcium intake of 800 mg without excessive urine losses. The kidney stone diet hinges on this combination. The articles show data linking urine calcium to diet calcium, as you asked about. You mention a diet sodium of 1200 mg, but does the 24 hour urine register so low a value – 52 meq? If indeed you have so low a urine sodium and also high urine calcium I would consider uncommon underlying reasons. Regards, Fred Coe
Dr Coe, I forgot to add to my earlier email today that as of last year I do not have Primary Hyperparathyroidism, it’s been tested several times and my numbers are fine.
Hi Carol, An important point. I presume you mean your fasting, morning serum calcium is 10.0 or less on several occasions. Fred
Hello Dr Coe.
I’ve read so many studies on how phytate (IP6) helps with kidney stones and bone chemistry.
One recent study I found below shows how phytate reduces urinary calcium in patients,
Do you think this study is compelling and well constructed.
Thanks
https://link.springer.com/content/pdf/10.1007/s00240-022-01357-8.pdf?pdf=button%20sticky
Hi Jonathan, It is a nice study. I would not use the material for you right now – too much uncertainty. Fred
Hi! I am a 69 yr old female with a pth of 98 with normal blood calcium but 356 24 hour urine calcium. I have low 20 Vitamin D and had blood pressure emergency of 190/109. I am on .37 levothyroxine for 45 years having had radioactive isotopes. I have normal bone scan. Wondering if I can do anything to avoid parathyroid surgery. Thank you.
Hi Marcia, When you say ‘normal’ blood calcium you mean below 10 mg/dl, I presume. Lets assume that is true. High PTH would then most reflect the low vitamin D level, low calcium diet, or perhaps mild reduction of kidney function. The last is reasonable given you have severe hypertension. But the high urine calcium is not consistent with low vitamin D or low calcium diet or reduced kidney function. This brings me back to the serum calcium. It is crucial what the values were. I am suspicious they are around 10 mg/dl and if so PHPT is a reasonable explanation. Regards, Fred Coe
Dear Colleague, I still do not understand why the normal range are always referred to mg or mmil/24H.
I use always 24H metabolic urinary collection but, are the normal range of ureic nitrogen, cytraturia, calcium, phosphorus, magnesium, sodium, kalium, ossalate referred to a patient with 1,73 s.m. body area? I didn’t find any indication in literature.
Usually I believe that it doesn’t exist more standardized evaluation but precision or personalized value for each one different patients.
What do you think about? This is mandatory e.g. for calculating protein intake by Urea Nitrogen Appearance or interviewing patient about your various different foods/day for each one component above descripted.
Final, the various urinary component, especially in kidney frequent stone former patient, it is necessary to consider the kidney ageing and I believe could be different for above parameters, like kidney function. In fact, at 70’s it could be considered normal a GFR about 70 mL/min/normalized body surface.
Thanks to read me, I hope you suggest me your opinion because I would work for a paper on 70 kidney frequent stone patients using math normalization for weight or body surface.
Hello Professor Bolasco, The 24 hour urine for stone formers aims at urine conditions causing stones not at mineral physiology, so normalization by BSA is not helpful. As for aging, urine calcium – and calcium stones – fall with age, but stone 24 hour urines are not aimed at assaying aging. So we agree but given the different uses of the urine chemistries normalization would have no benefits. Regards, Fred Coe